

การหาลำดับ mRNA แบบเต็มความยาว -PacBio





ข้อดีของการบริการ
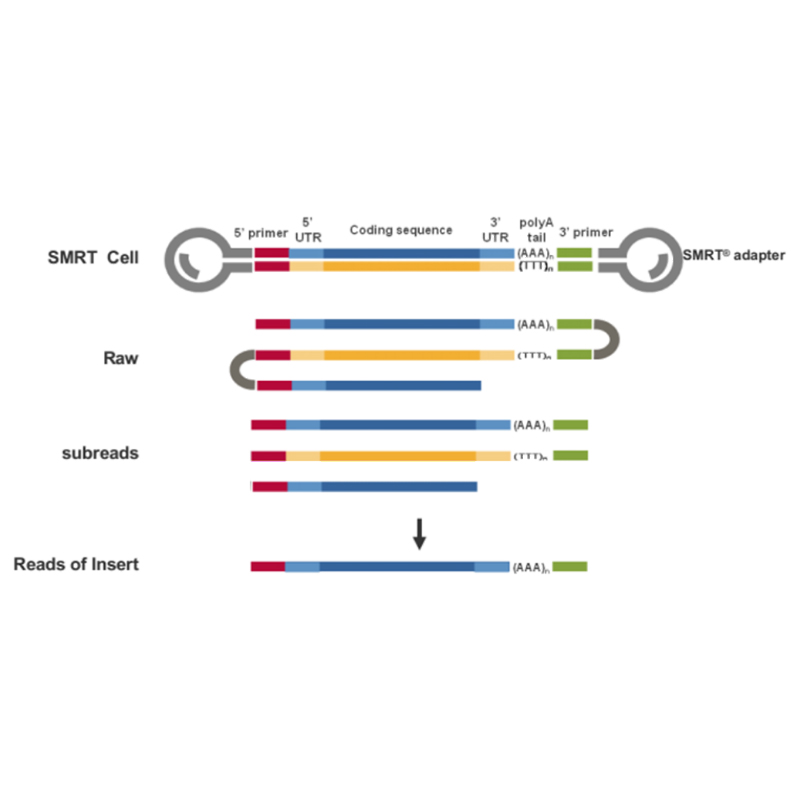
● อ่านค่าโมเลกุล cDNA แบบเต็มความยาวได้โดยตรงจากปลาย 3' ถึงปลาย 5'
● ความละเอียดระดับรูปแบบ ISO ในโครงสร้างลำดับ
● การถอดเสียงที่มีความแม่นยำและเที่ยงตรงสูง
● เข้ากันได้สูงกับสายพันธุ์ vaiours
● ความสามารถในการจัดลำดับขนาดใหญ่ด้วยการติดตั้งแพลตฟอร์มการจัดลำดับ PacBio Sequel II จำนวน 4 ชุด
● มีประสบการณ์สูงกับโครงการหาลำดับ RNA ที่ใช้ Pacbio มากกว่า 700 โครงการ
● การส่งมอบผลลัพธ์ตาม BMKCloud: การทำเหมืองข้อมูลแบบกำหนดเองพร้อมใช้งานบนแพลตฟอร์ม
● บริการหลังการขายจะมีอายุการใช้งาน 3 เดือนเมื่อโครงการเสร็จสิ้น
ข้อมูลจำเพาะของบริการ
แพลตฟอร์ม: PacBio ภาคต่อ II
ไลบรารีลำดับ: ไลบรารี mRNA ที่ได้รับการเสริมสมรรถนะด้วย Poly A
ปริมาณข้อมูลที่แนะนำ: 20 Gb/ตัวอย่าง (ขึ้นอยู่กับสายพันธุ์)
FLNC (%): ≥75%
*FLNC: ทรานสคริปต์ที่ไม่ใช่แบบไคเมอริกแบบเต็มความยาว
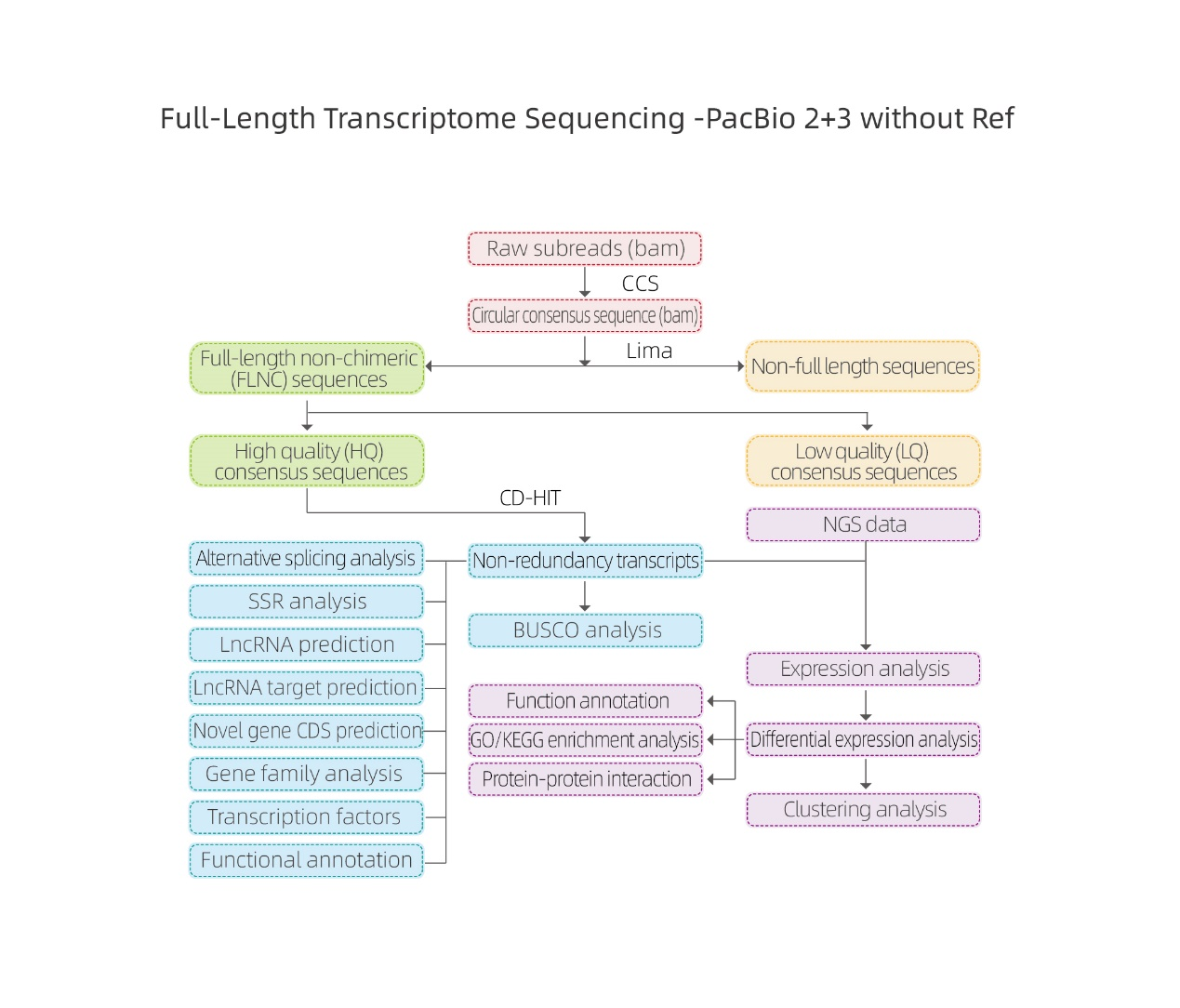
การวิเคราะห์ทางชีวสารสนเทศ
● การประมวลผลข้อมูลดิบ
● บัตรประจำตัวการถอดเสียง
● โครงสร้างลำดับ
● ปริมาณนิพจน์
● คำอธิบายประกอบฟังก์ชัน
ข้อกำหนดตัวอย่างและการจัดส่ง
ข้อกำหนดตัวอย่าง:
นิวคลีโอไทด์:
| ความเข้มข้น(ng/μl) | ปริมาณ (ไมโครกรัม) | ความบริสุทธิ์ | ความซื่อสัตย์ |
| ≥ 120 | ≥ 0.6 | OD260/280=1.7-2.5 โอดี260/230=0.5-2.5 มีการปนเปื้อนของโปรตีนหรือ DNA อย่างจำกัดหรือไม่มีเลยบนเจล | สำหรับพืช: RIN≥7.5; สำหรับสัตว์: RIN≥8.0; 5.0≥ 28S/18S≥1.0; ระดับความสูงพื้นฐานที่จำกัดหรือไม่มีเลย |
กระดาษทิชชู: น้ำหนัก(แห้ง):≥1ก
*สำหรับเนื้อเยื่อที่มีขนาดเล็กกว่า 5 มก. เราแนะนำให้ส่งตัวอย่างเนื้อเยื่อแช่แข็งแบบแฟลช (ในไนโตรเจนเหลว)
การระงับเซลล์:จำนวนเซลล์ = 3×106- 1×107
*เราแนะนำให้จัดส่งไลเซทเซลล์แช่แข็งในกรณีที่เซลล์นั้นนับน้อยกว่า 5×105แนะนำให้ใช้แฟลชแช่แข็งในไนโตรเจนเหลว ซึ่งเหมาะสำหรับการสกัดแบบไมโคร
ตัวอย่างเลือด:ปริมาตร≥1มล
จุลินทรีย์:มวล ≥ 1 ก
การจัดส่งตัวอย่างที่แนะนำ
คอนเทนเนอร์:
หลอดหมุนเหวี่ยงขนาด 2 มล. (ไม่แนะนำให้ใช้ฟอยล์ดีบุก)
การติดฉลากตัวอย่าง: กลุ่ม+ทำซ้ำ เช่น A1, A2, A3;บี1 บี2 บี3......
การจัดส่ง:
1. น้ำแข็งแห้ง: ตัวอย่างจะต้องบรรจุในถุงและฝังในน้ำแข็งแห้ง
2. หลอด RNAstable: ตัวอย่าง RNA สามารถทำให้แห้งในหลอดรักษาเสถียรภาพ RNA (เช่น RNAstable®) และจัดส่งในอุณหภูมิห้อง
ขั้นตอนการทำงานบริการ

การออกแบบการทดลอง

การจัดส่งตัวอย่าง

การสกัดอาร์เอ็นเอ

การก่อสร้างห้องสมุด

การเรียงลำดับ

การวิเคราะห์ข้อมูล

บริการหลังการขาย
1.การกระจายความยาว FLNC
ความยาวของการอ่านที่ไม่ใช่แบบคิเมริกแบบเต็มความยาว (FLNC) ระบุความยาวของ cDNA ในการสร้างห้องสมุดการกระจายความยาว FLNC เป็นตัวบ่งชี้สำคัญในการประเมินคุณภาพการก่อสร้างห้องสมุด
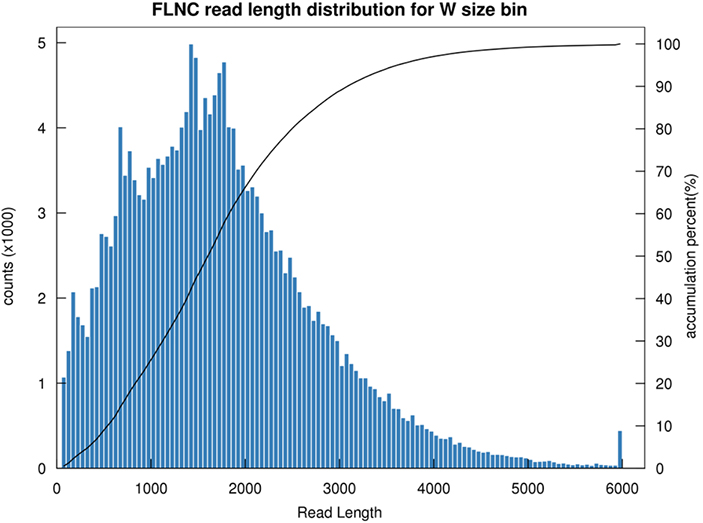
การกระจายความยาวการอ่าน FLNC
2. การกระจายความยาวภูมิภาค ORF ให้สมบูรณ์
เราใช้ TransDecoder เพื่อทำนายบริเวณการเข้ารหัสโปรตีนและลำดับกรดอะมิโนที่เกี่ยวข้องเพื่อสร้างชุดยีนเดียว ซึ่งมีข้อมูลการถอดเสียงที่สมบูรณ์และไม่ซ้ำซ้อนในทุกตัวอย่าง
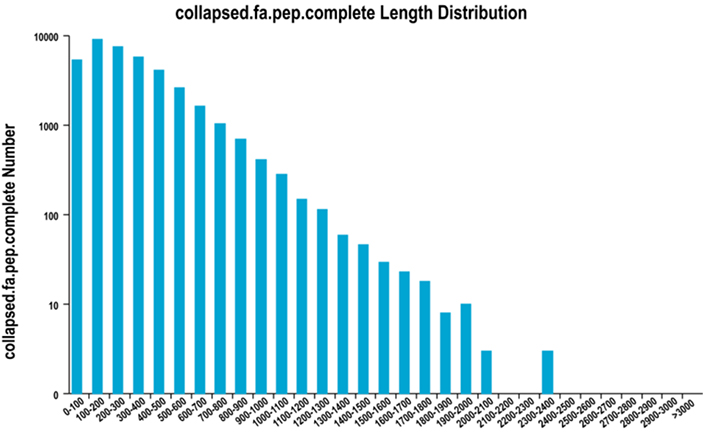
การกระจายความยาวขอบเขต ORF ให้สมบูรณ์
3.การวิเคราะห์การเพิ่มคุณค่าวิถี KEGG
การถอดเสียงที่แสดงความแตกต่าง (DET) สามารถระบุได้โดยการจัดข้อมูลการจัดลำดับ RNA ที่ใช้ NGS ให้สอดคล้องกับชุดการถอดเสียงแบบเต็มความยาวที่สร้างโดยข้อมูลการจัดลำดับ PacBioDET เหล่านี้สามารถประมวลผลเพิ่มเติมสำหรับการวิเคราะห์เชิงฟังก์ชันต่างๆ เช่น การวิเคราะห์การเพิ่มคุณค่าวิถี KEGG
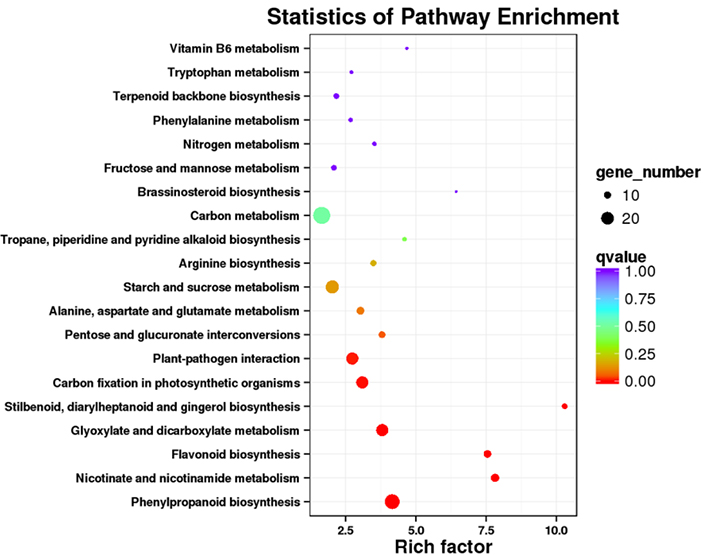
การเพิ่มคุณค่าวิถี DET KEGG - พล็อตจุด
คดีบีเอ็มเค
พลวัตการพัฒนาของทรานสคริปโตมต้นกำเนิด Populus
ที่ตีพิมพ์: วารสารเทคโนโลยีชีวภาพพืช, 2019
กลยุทธ์การจัดลำดับ:
การเก็บตัวอย่าง:บริเวณต้นกำเนิด: เอเพ็กซ์, ปล้องแรก (IN1), ปล้องที่สอง (IN2), ปล้องที่สาม (IN3), ปล้อง (IN4) และปล้องปล้อง (IN5) จาก Nanlin895
ลำดับ NGS:RNA ของบุคคล 15 คนถูกรวมเข้าด้วยกันเป็นตัวอย่างทางชีวภาพหนึ่งตัวอย่างการจำลองทางชีวภาพสามรายการของแต่ละจุดได้รับการประมวลผลสำหรับลำดับ NGS
ลำดับ TGS:บริเวณลำต้นถูกแบ่งออกเป็นสามบริเวณ ได้แก่ ปลาย IN1-IN3 และ IN4-IN5แต่ละภูมิภาคได้รับการประมวลผลสำหรับการเรียงลำดับ PacBio ด้วยไลบรารีสี่ประเภท: 0-1 kb, 1-2 kb, 2-3 kb และ 3-10 kb
ผลลัพธ์ที่สำคัญ
1. มีการระบุสำเนาฉบับเต็มทั้งหมด 87,150 รายการ โดยระบุไอโซฟอร์มใหม่ 2,081 รายการ และไอโซฟอร์มต่อประสานทางเลือกใหม่ 62,058 รายการ
ระบุยีนฟิวชั่น 2.1187 lncRNA และ 356 ตัว
3. จากการเติบโตขั้นปฐมภูมิไปจนถึงการเติบโตขั้นทุติยภูมิ มีการระบุการถอดเสียงที่แสดงออกแตกต่างกัน 15,838 รายการจากยีนที่แสดงออกแตกต่างกัน 995 ยีนใน DEG ทั้งหมด 1,216 รายการเป็นปัจจัยการถอดรหัส ซึ่งส่วนใหญ่ยังไม่มีการรายงาน
การวิเคราะห์การเพิ่มคุณค่า 4.GO เปิดเผยความสำคัญของการแบ่งเซลล์และกระบวนการลดออกซิเดชันในการเจริญเติบโตขั้นปฐมภูมิและทุติยภูมิ
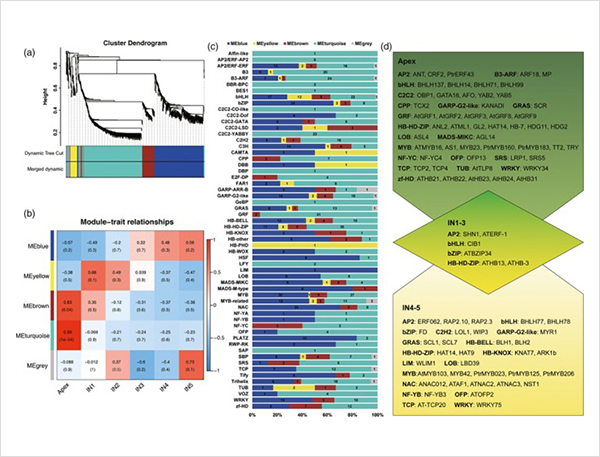
เหตุการณ์การต่อรอยทางเลือกและไอโซฟอร์มที่แตกต่างกัน
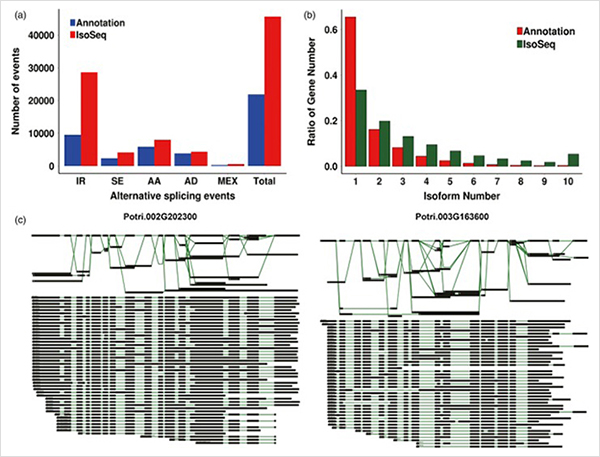
การวิเคราะห์ WGCNA เกี่ยวกับปัจจัยการถอดรหัส
อ้างอิง
Chao Q, Gao ZF, Zhang D และอื่น ๆพลวัตการพัฒนาของทรานสคริปโตมต้นกำเนิด Populusพืชเทคโนโลยีชีวภาพ J. 2019;17(1):206-219.ดอย:10.1111/pbi.12958





