

Genotypning med hög genomströmning, särskilt på storskalig population, är ett grundläggande steg i genetiska associationsstudier, vilket ger genetisk grund för funktionell genupptäckt, evolutionär analys, etc. Istället för djup omsekvensering av hela genomet, reducerad representationsgenomsekvensering (RRGS) ) introduceras för att minimera sekvenseringskostnaden per prov, samtidigt som rimlig effektivitet vid upptäckt av genetisk markör upprätthålls.Detta uppnås vanligtvis genom att extrahera restriktionsfragment inom ett givet storleksintervall, som kallas reducerat representationsbibliotek (RRL).Specific-locus amplified fragment sequencing (SLAF-Seq) är en egenutvecklad strategi för de novo SNP-upptäckt och SNP-genotypning av stora populationer.
Tekniskt arbetsflöde
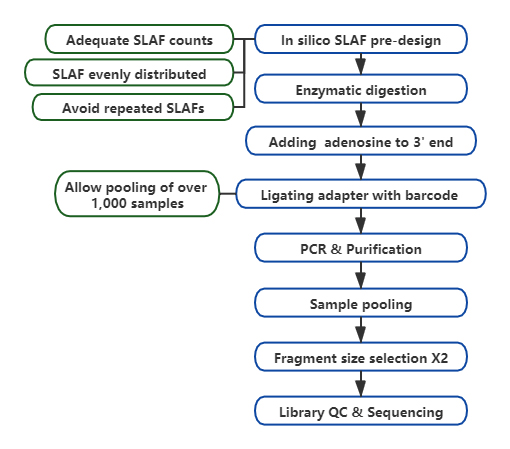
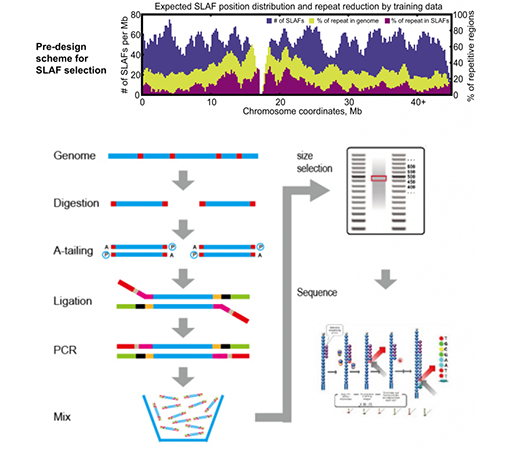
SLAF vs existerande RRL-metoder
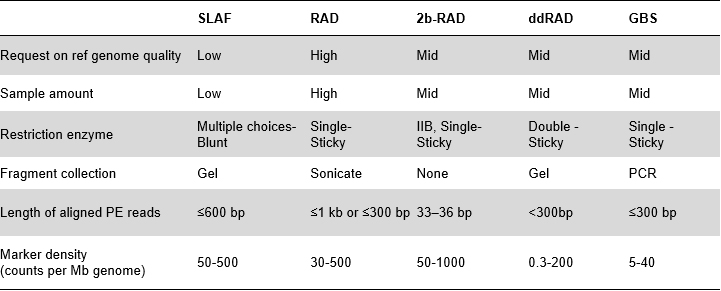
Fördelar med SLAF
Högre effektivitet för upptäckt av genetiska markörer– I kombination med sekvenseringsteknik med hög genomströmning kan SLAF-Seq uppnå hundratusentals taggar som upptäckts inom hela genomet för att uppfylla kraven från olika forskningsprojekt, antingen med eller utan ett referensgenom.
Anpassad och flexibel experimentell design– För olika forskningsmål eller arter finns olika enzymatiska matsmältningsstrategier tillgängliga inklusive enkel-enzym, dubbel-enzym och multi-enzym matsmältning.Matsmältningsstrategin kommer att förutvärderas i silico för att säkerställa en optimal enzymdesign.
Hög effektivitet i enzymatisk matsmältning– Fördesignad enzymatisk matsmältning ger mer jämnt fördelade SLAF på kromosomen.Fragmentuppsamlingseffektiv kan uppnå över 95 %.
Undvik upprepade sekvenser– Procentandelen av repetitiva sekvenser i SLAF-Seq-data reduceras till lägre än 5 %, särskilt i arter med hög nivå av repetitiva element, såsom vete, majs, etc.
Egenutvecklat bioinformatiskt arbetsflöde– BMK utvecklade ett integrerat bioinformatiskt arbetsflöde som är tillämpligt på SLAF-Seq-teknik för att säkerställa tillförlitlighet och noggrannhet i slutresultatet.
Tillämpning av SLAF
Genetisk kopplingskarta
Genetisk kartkonstruktion med hög densitet och identifiering av loci som kontrollerar egenskaper av blomtyp i Chrysanthemum (Chrysanthemum x morifolium Ramat.)
Tidskrift: Horticulture Research Publicerad: 2020.7

GWAS
Identifiering av en kandidatgen associerad med isofavoninnehåll i sojabönsfrön med hjälp av genomomfattande association och länkkartering
Journal: the Plant Journal Publicerad: 2020.08

Evolutionär genetik
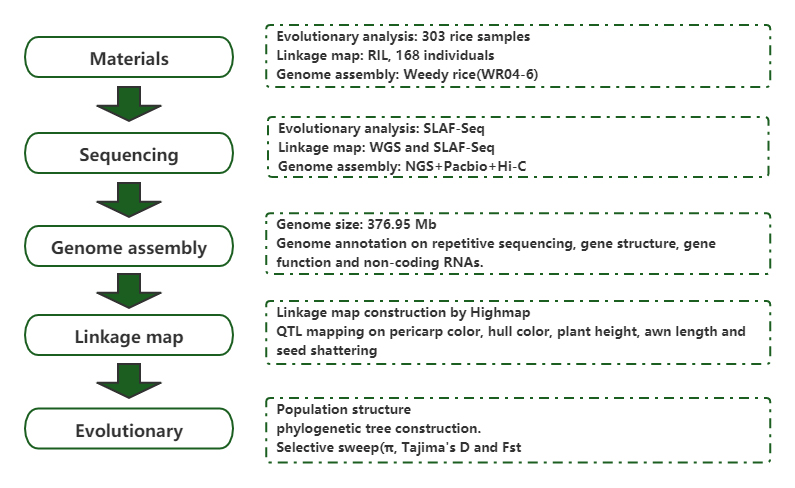
Populationsgenomanalys och de novo-sammansättning avslöjar ursprunget till ogräsris som ett evolutionärt spel
Tidskrift: Molecular Plant Publicerad: 2019.5
Bulk Segregant Analysis (BSA)
GmST1, som kodar för ett sulfotransferas, ger resistens mot sojabönmosaikvirusstammarna G2 och G3
Tidskrift: Plant, Cell&Environment Publicerad: 2021.04

Referens
Sun X, Liu D, Zhang X, et al.SLAF-Seq: en effektiv metod för storskalig de novo SNP-upptäckt och genotypning med hjälp av sekvensering med hög genomströmning[J].Plos ett, 2013, 8(3):e58700
Song X, Xu Y, Gao K, et al.Genetisk kartkonstruktion med hög täthet och identifiering av loci som kontrollerar egenskaper av blomtyp i krysantemum (Chrysanthemum × morifolium Ramat.).Hortic Res.2020;7:108.
Wu D, Li D, Zhao X, et al.Identifiering av en kandidatgen associerad med isoflavoninnehåll i sojabönsfrön med hjälp av genomomfattande association och länkkartering.Plant J. 2020;104(4): 950-963.
Sun J, Ma D, Tang L, et al.Populationsgenomisk analys och De Novo Assembly avslöjar ursprunget till Weedy Rice som ett evolutionärt spel.Mol Plant.2019;12(5):632-647.Mol Plant.2018;11(11):1360-1376.
Zhao X, Jing Y, Luo Z, et al.GmST1, som kodar för ett sulfotransferas, ger resistens mot sojabönmosaikvirusstammarna G2 och G3.Växtcellsmiljö.2021;10.1111/st.14066
Posttid: Jan-04-2022