

Hi-C-baserad Genome Assembly







Servicefördelar
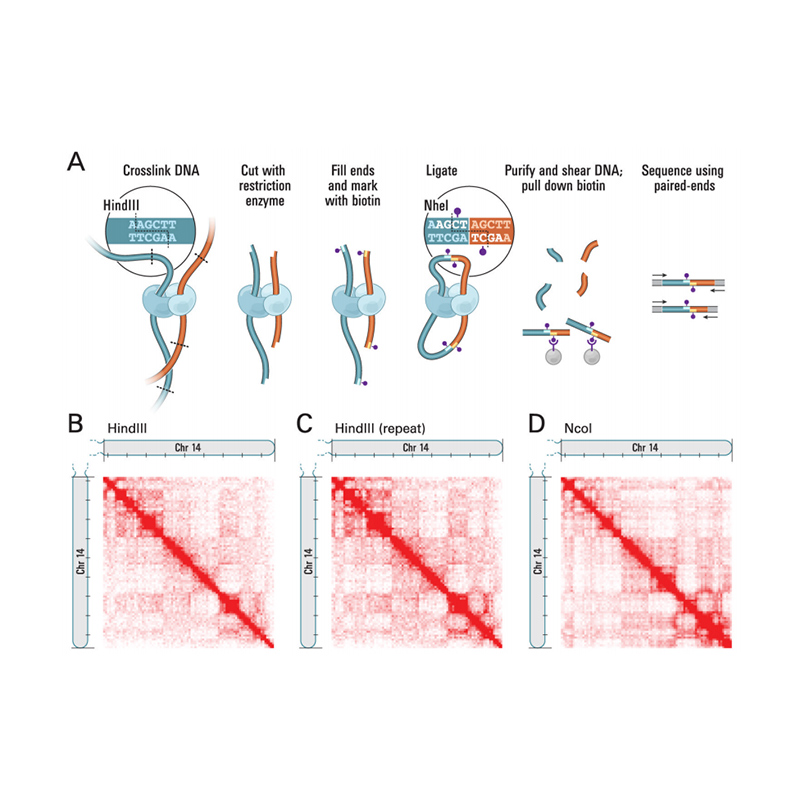
Översikt över Hi-C
(Lieberman-Aiden E et al.,Vetenskap, 2009)
● Inget behov av att konstruera genetisk population för kontig förankring;
● Högre markördensitet som leder till högre förankringsförhållande för contigs vid över 90 %;
● Möjliggör utvärdering och korrigeringar av befintliga genomsamlingar;
● Kortare omloppstid med högre noggrannhet vid genomsamling;
● Riklig erfarenhet med över 1 000 Hi-C-bibliotek konstruerade för över 500 arter;
● Över 100 framgångsrika fall med en ackumulerad publicerad effektfaktor på över 760;
● Hi-C-baserad genomsammansättning för polyploid genom, 100 % förankringsgrad uppnåddes i tidigare projekt;
● Interna patent och upphovsrätt till programvara för Hi-C-experiment och dataanalys;
● Egenutvecklad mjukvara för visualiserad datajustering, möjliggör manuell blockflyttning, reversering, återkallelse och omställning.
Servicespecifikationer
|
Bibliotekstyp
|
Plattform | Läs Längd | Rekommendera strategi |
| Hi-C | Illumina NovaSeq | PE150 | ≥ 100X |
Bioinformatikanalyser
● Kvalitetskontroll av rådata
● Hi-C-bibliotekets kvalitetskontroll
● Hi-C-baserad genomsammansättning
● Utvärdering efter montering
Exempel på krav och leverans
Exempelkrav:
| Djur | Svamp | Växter
|
| Fryst vävnad: 1-2g per bibliotek Celler: 1x 10^7 celler per bibliotek | Fryst vävnad: 1g per bibliotek | Fryst vävnad: 1-2g per bibliotek
|
| *Vi rekommenderar starkt att du skickar minst 2 alikvoter (1 g vardera) för Hi-C-experimentet. | ||
Rekommenderad provleverans
Behållare: 2 ml centrifugrör (tunnfolie rekommenderas inte)
För de flesta prover rekommenderar vi att inte konservera i etanol.
Provmärkning: Proverna måste vara tydligt märkta och identiska med det inlämnade provinformationsformuläret.
Försändelse: Torris: Proverna måste först packas i påsar och grävas ner i torris.
Service arbetsflöde

Experimentdesign

Provleverans

DNA-extraktion

Byggande av bibliotek

Sekvensering

Dataanalys

Service efter försäljning
*Demoresultat som visas här är alla från genom publicerade med Biomarker Technologies
1.Hi-C interaktion värmekarta överCamptotheca acuminatagenomet.Som visas på kartan är intensiteten av interaktioner negativt korrelerad med det linjära avståndet, vilket indikerar en mycket noggrann sammansättning på kromosomnivå.(Förankringsgrad: 96,03 %)
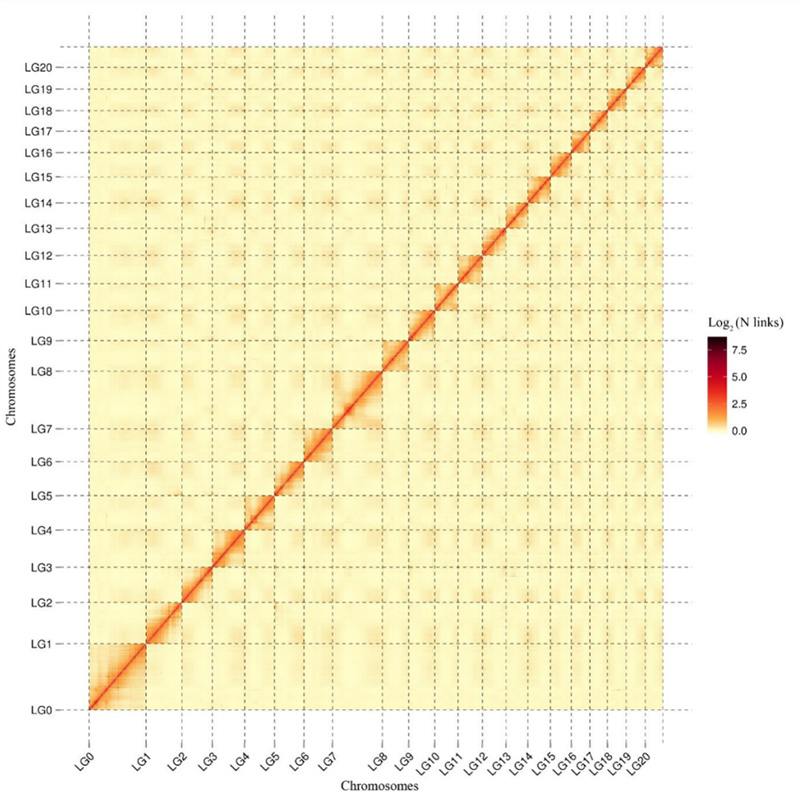
Kang M et al.,Naturkommunikation, 2021
2.Hi-C underlättade valideringen av inversioner mellanGossypium hirsutumL. TM-1 A06 ochG. arboreumChr06
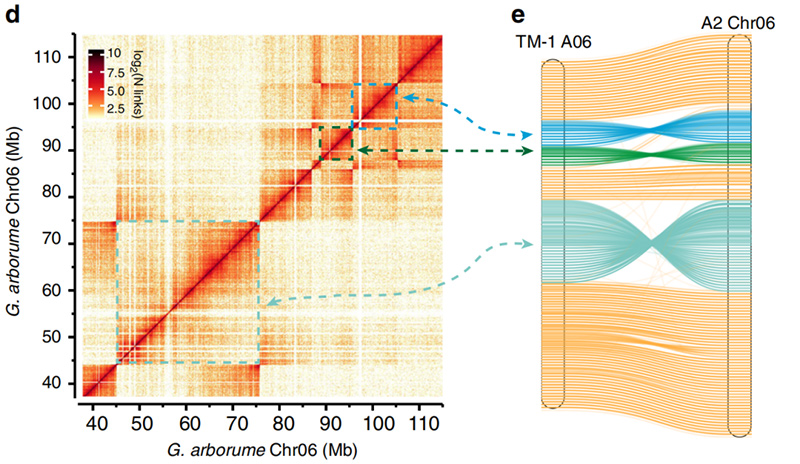
Yang Z et al.,Naturkommunikation, 2019
3. Montering och biallelisk differentiering av kassava-genomet SC205.Hi-C värmekarta visar tydlig splittring i homologa kromosomer.
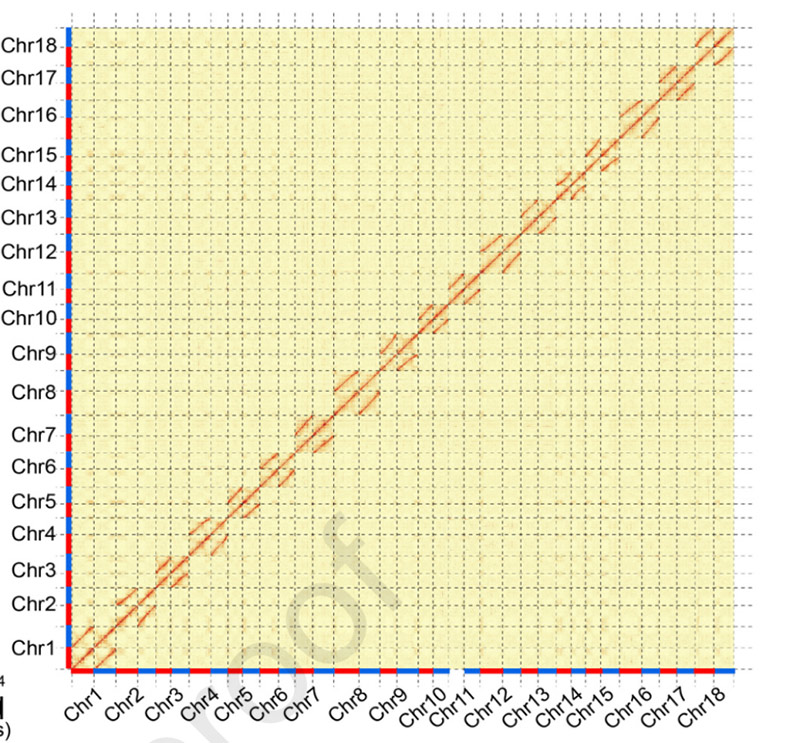
Hu W et al.,Molekylär växt, 2021
4.Hi-C värmekarta på två Ficus-arter genom montering:F.microcarpa(förankringsgrad: 99,3%) ochF.hispida (förankringsförhållande: 99,7 %)
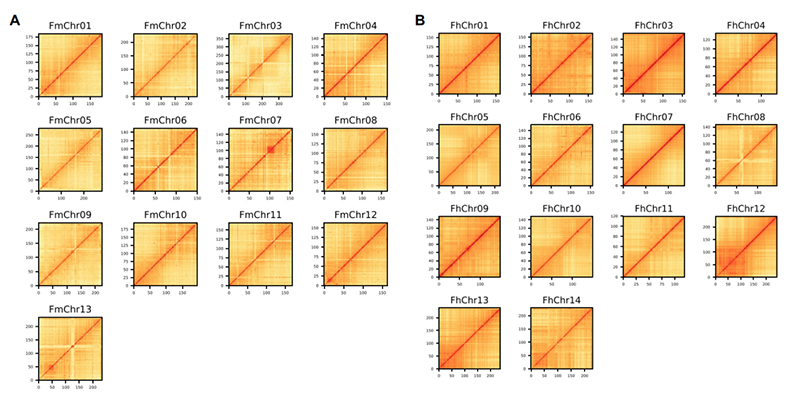
Zhang X et al.,Cell, 2020
BMK Fall
Genomer från Banyanträdet och pollinatorgetingen ger insikter i fikongetingens samutveckling
Publicerad: Cell, 2020
Sekvenseringsstrategi:
F. microcarpa genom: Ca.84 X PacBio RSII (36,87 Gb) + Hi-C (44 Gb)
F. hispidagenom: Ca.97 X PacBio RSII (36,12 Gb) + Hi-C (60 Gb)
Eupristina verticillatagenom: Ca.170 X PacBio RSII (65 Gb)
Nyckelresultat
1. Två banyanträdgenom och en pollinatorgetinggenom konstruerades med hjälp av PacBio-sekvensering, Hi-C och länkkarta.
(1)F. microcarpagenom: En samling på 426 Mb (97,7 % av uppskattad genomstorlek) etablerades med contig N50 på 908 Kb, BUSCO-poäng på 95,6 %.Totalt 423 Mb sekvenser förankrades till 13 kromosomer med Hi-C.Genomannotering gav 29 416 proteinkodande gener.
(2)F. Hispidagenom: En sammansättning på 360 Mb (97,3 % av uppskattad genomstorlek) gav ett utbyte med contig N50 på 492 Kb och BUSCO-poäng på 97,4 %.Totalt 359 Mb-sekvenser förankrades på 14 kromosomer med Hi-C och mycket identiska med högdensitetslänkningskarta.
(3)Eupristina verticillatagenom: En samling på 387 Mb (uppskattad genomstorlek: 382 Mb) etablerades med contig N50 på 3,1 Mb och BUSCO-poäng på 97,7%.
2. Jämförande genomikanalys visade ett stort antal strukturvariationer mellan tvåFicusgenom, som gav ovärderlig genetisk resurs för adaptiva evolutionsstudier.Denna studie, för första gången, gav insikter i Fig-geting samevolution på genomisk nivå.
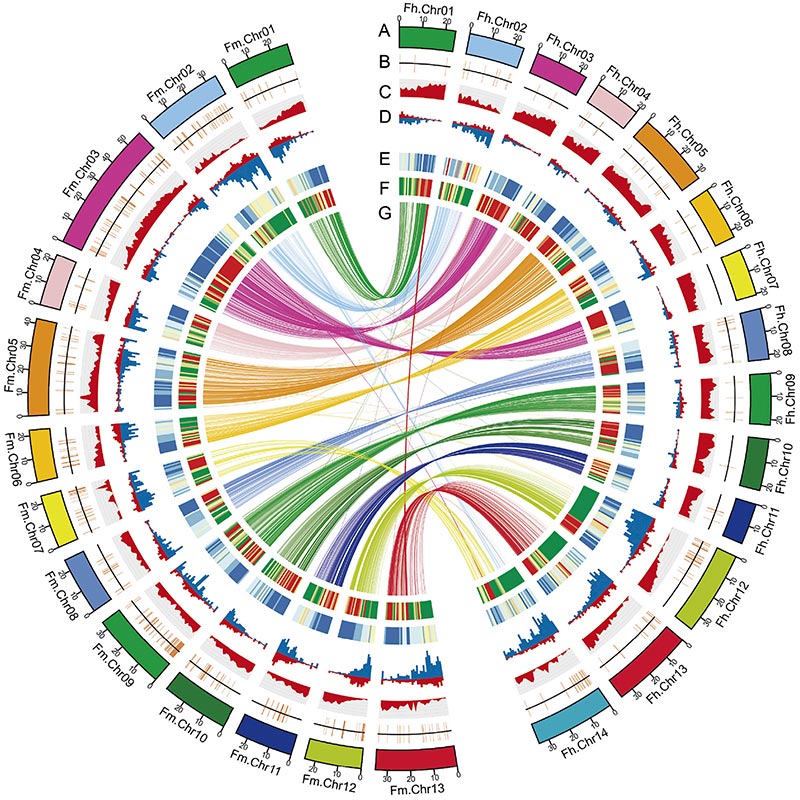 Circos diagram på genomiska egenskaper hos tvåFicusgenom, inklusive kromosomer, segmentella duplikationer (SD), transposoner (LTR, TE, DNA TE), genuttryck och synteny | 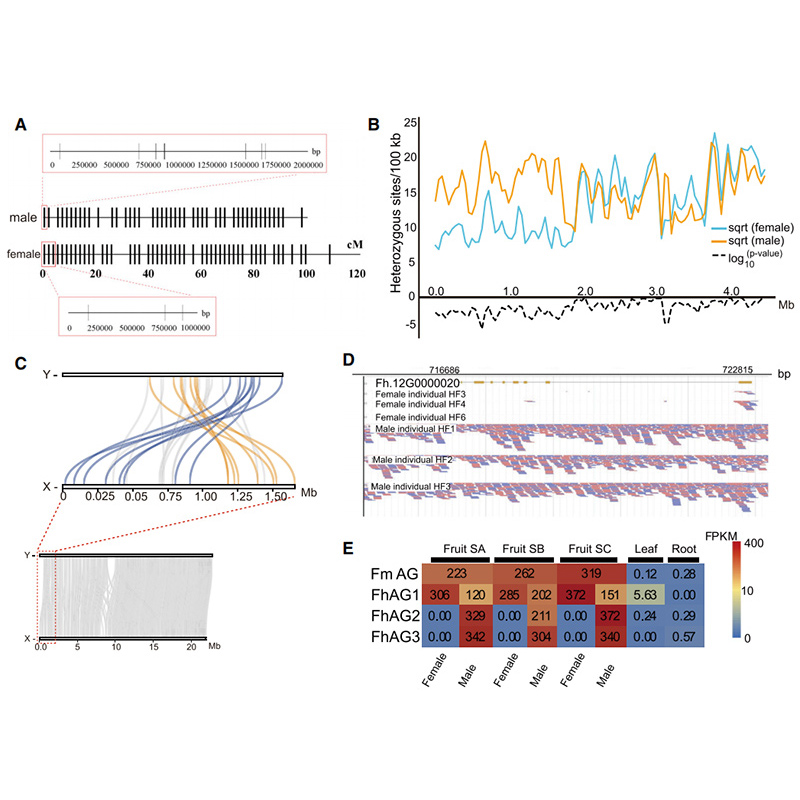 Identifiering av Y-kromosom- och könsbestämningskandidatgenen |
Zhang, X., et al."Genomen från Banyanträdet och pollinatorgetingen ger insikter i fikongetingens samutveckling."Cell 183.4(2020).





