

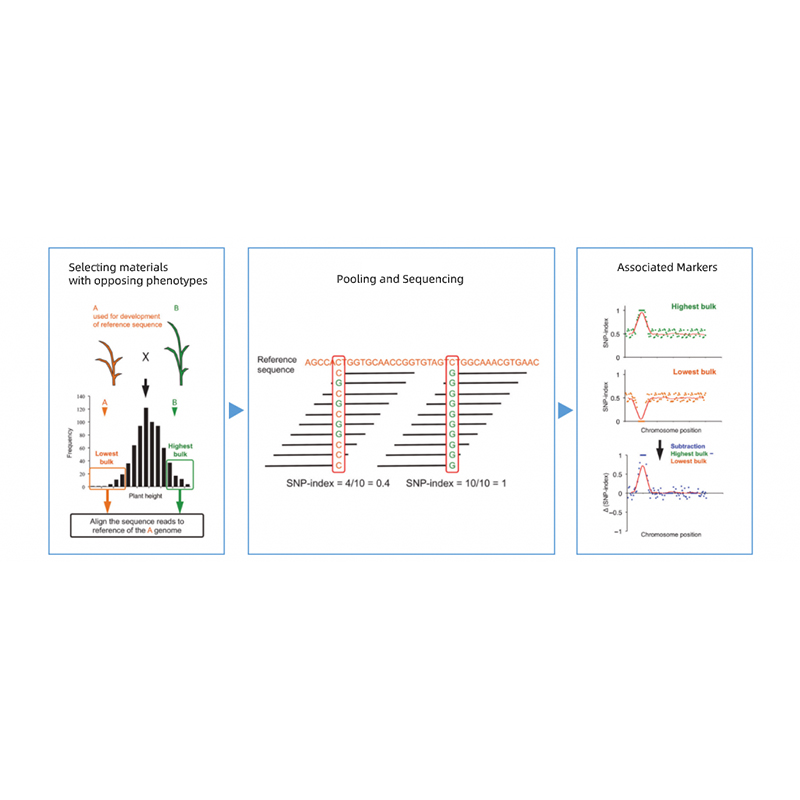
Bulk Segregant analys






Servicefördelar
Takagi et al., The plant journal, 2013
● Exakt lokalisering: Blandning av bulks med 30+30 till 200+200 individer för att minimera bakgrundsljud;icke-synonyma mutatanter-baserade kandidatregionförutsägelser.
● Omfattande analys: Fördjupad annotering av kandidatgenfunktioner, inklusive NR, SwissProt, GO, KEGG, COG, KOG, etc.
● Snabbare handläggningstid: Snabb genlokalisering inom 45 arbetsdagar.
● Lång erfarenhet: BMK har bidragit till tusentals lokalisering av egenskaper, som täcker olika arter som grödor, vattenlevande produkter, skog, blommor, frukter, etc.
Servicespecifikationer
Befolkning:
Segregering av avkomma till föräldrar med motsatta fenotyper.
t.ex. F2-avkomma, Backcrossing (BC), Rekombinant inavlad linje (RIL)
Blandningspool
För kvalitativa egenskaper: 30 till 50 individer (minst 20)/bulk
För kvantitativa tratis: topp 5% till 10% individer med antingen extrema fenotyper i hela populationen (minst 30+30).
Rekommenderat sekvenseringsdjup
Minst 20X/förälder och 1X/avkomma individ (t.ex. för avkommans blandning av 30+30 individer, kommer sekvenseringsdjupet att vara 30X per bulk)
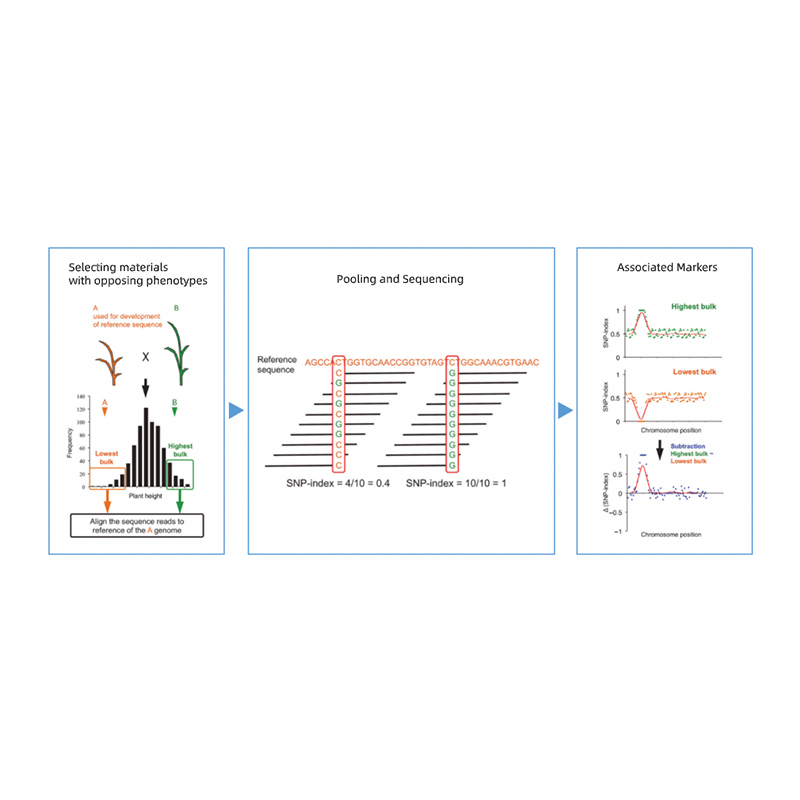
Bioinformatikanalyser
● Resekvensering av hela genomet
● Databehandling
● SNP/Indel-anrop
● Screening av kandidatregioner
● Annotering av kandidatgenfunktion
Exempel på krav och leverans
Exempelkrav:
Nukleotider:
| gDNA-prov | Vävnadsprov |
| Koncentration: ≥30 ng/μl | Växter: 1-2 g |
| Mängd: ≥2 μg (Volym ≥15 μl) | Djur: 0,5-1 g |
| Renhet: OD260/280= 1,6-2,5 | Helblod: 1,5 ml |
Service Arbetsflöde

Experimentdesign

Provleverans

RNA-extraktion

Byggande av bibliotek

Sekvensering

Dataanalys

Service efter försäljning
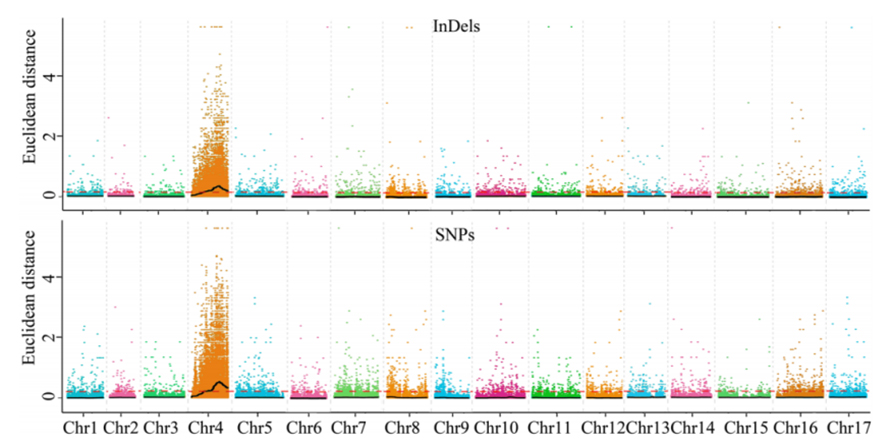
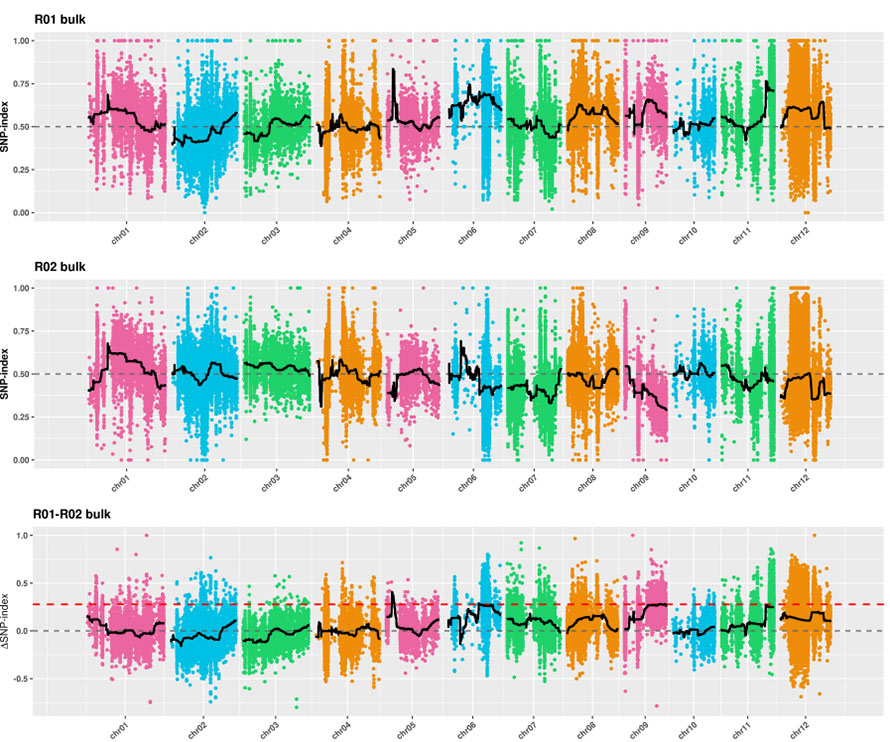
1.Associationsanalys baseras på euklidisk distans (ED) för att identifiera kandidatregion.I följande figur
X-axel: Kromosomnummer;Varje punkt representerar ett ED-värde för en SNP.Den svarta linjen motsvarar det inpassade ED-värdet.Ett högre ED-värde indikerar ett mer signifikant samband mellan platsen och fenotypen.Röd streckad linje representerar tröskeln för signifikant association.
2.Associationsanalys baserad utan SNP-index
X-axel: Kromosomnummer;Varje punkt representerar SNP-indexvärde.Den svarta linjen står för monterat SNP-indexvärde.Ju större värdet är, desto mer betydelsefull är föreningen.
BMK Fall
Major-effekten kvantitativa egenskapen locus Fnl7.1 kodar för ett rikligt protein i sen embryogenes associerat med frukthalsens längd i gurka
Publicerad: Plant Biotechnology Journal, 2020
Sekvenseringsstrategi:
Föräldrar (Jin5-508, YN): Helgenomsekvensering för 34× och 20×.
DNA-pooler (50 långhalsade och 50 korthalsade): Återsekvensering för 61× och 52×
Nyckelresultat
I denna studie genererades segregerande population (F2 och F2:3) genom att korsa långhalsad gurkalinje Jin5-508 och korthalsad YN.Två DNA-pooler konstruerades av 50 extremt långhalsade individer och 50 extrema korthalsade individer.Major-effekt QTL identifierades på Chr07 genom BSA-analys och traditionell QTL-kartläggning.Kandidatregionen minskades ytterligare genom finkartläggning, kvantifiering av genuttryck och transgena experiment, som avslöjade nyckelgenen för att kontrollera nacklängden, CsFnl7.1.Dessutom befanns polymorfism i CsFnl7.1-promotorregionen vara associerad med motsvarande uttryck.Ytterligare fylogenetisk analys antydde att Fnl7.1-lokus med stor sannolikhet kommer från Indien.
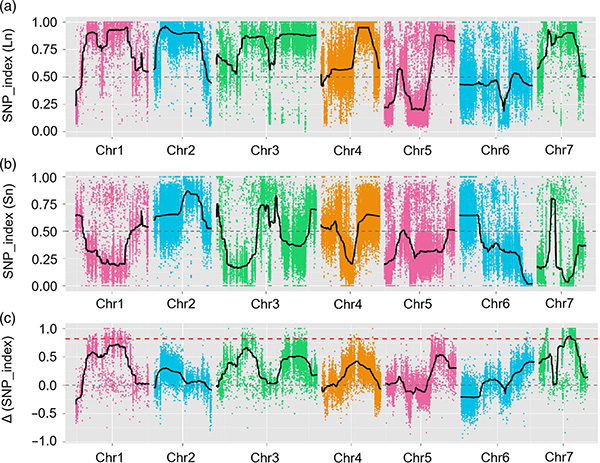 QTL-kartläggning i BSA-analys för att identifiera kandidatregion associerad med gurkans halslängd | 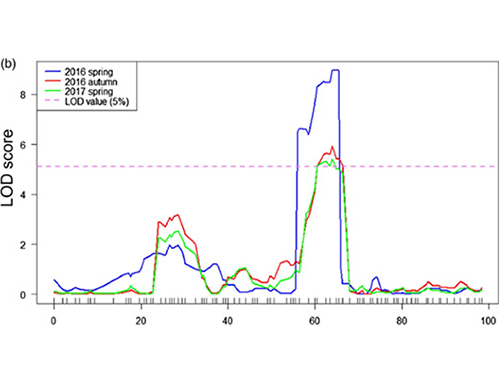 LOD-profiler av gurkhalslängd QTL identifierade på Chr07 |
Xu, X. et al."Major-effekten kvantitativa egenskapen locus Fnl7.1 kodar för ett sen embryogenes rikligt protein associerat med frukthalslängd i gurka."Plant Biotechnology Journal 18.7(2020).





