

Sequenciamento de Fragmentos Amplificados com Locus Específico (SLAF-Seq)





Detalhes do serviço
Esquema Técnico
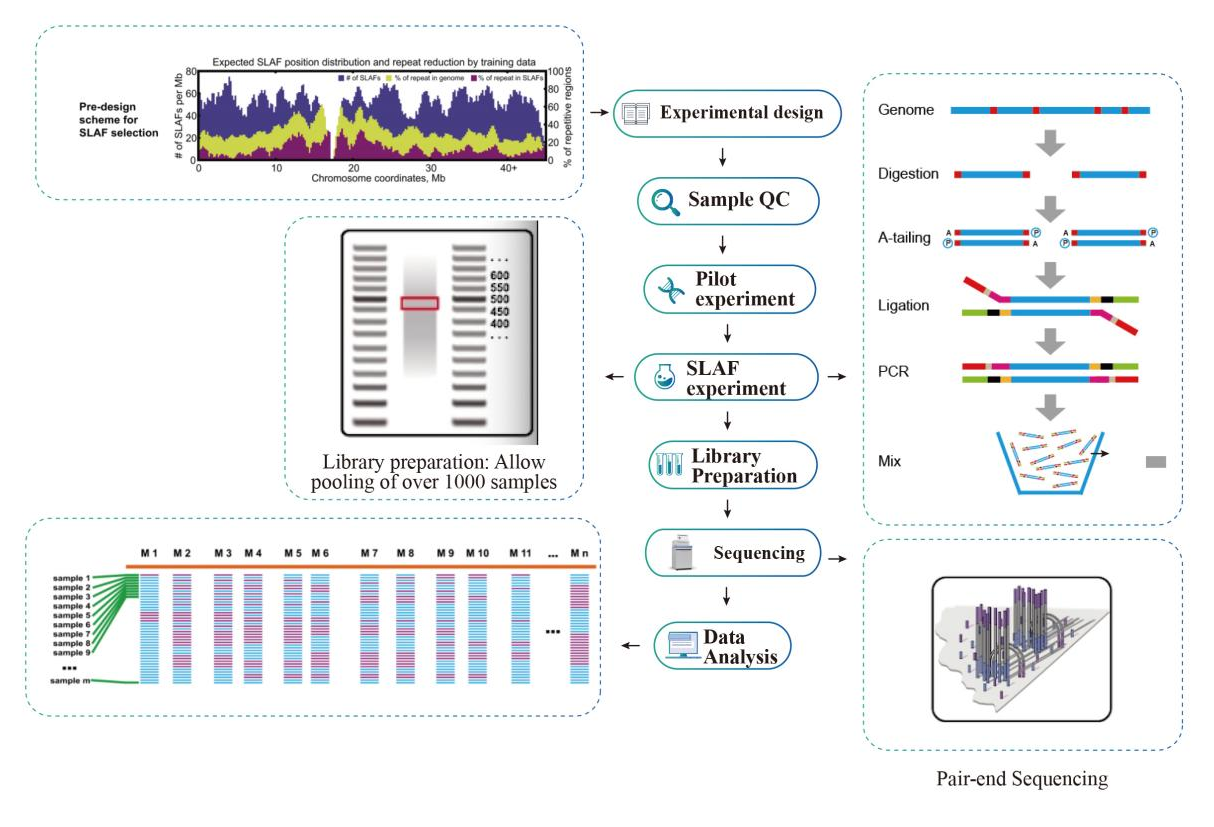
Fluxo de trabalho

Vantagens do serviço
Alta eficiência de descoberta de marcadores- A tecnologia de sequenciamento de alto rendimento auxilia o SLAF-Seq na descoberta de centenas de milhares de tags em todo o genoma.
Baixa dependência do genoma- Pode ser aplicado a espécies com ou sem genoma de referência.
Projeto de esquema flexível- Digestão de enzima única, enzima dupla, multienzima e vários tipos de enzimas, todas podem ser selecionadas para atender a diferentes objetivos ou espécies de pesquisa.A pré-avaliação in silico é usada para garantir um projeto enzimático ideal.
Digestão enzimática eficiente- O pré-experimento foi realizado para otimizar as condições, o que torna o experimento formal estável e confiável.A eficiência da coleta de fragmentos pode atingir mais de 95%.
Tags SLAF distribuídas uniformemente- As tags SLAF são distribuídas uniformemente em todos os cromossomos, atingindo uma média de 1 SLAF por 4 kb.
Evitar eficazmente repetições- A sequência repetitiva nos dados SLAF-Seq é reduzida para menos de 5%, especialmente em espécies com alto nível de repetições, como trigo, milho, etc.
Ampla experiência-Mais de 2.000 projetos SLAF-Seq fechados em centenas de espécies abrangendo plantas, mamíferos, pássaros, insetos, organismos aquáticos, etc.
Fluxo de trabalho de bioinformática autodesenvolvido- Um fluxo de trabalho de bioinformática integrado para SLAF-Seq foi desenvolvido pela BMKGENE para garantir confiabilidade e precisão do resultado final.
Especificações de serviço
| Plataforma | Conc.(ng/gl) | Total (ug) | OD260/280 |
| Illumina NovaSeq | >35 | >1.6(Volume>15μl) | 1,6-2,5 |
Estratégia de sequenciamento recomendada
Profundidade de sequenciamento: 10X/Tag
| Tamanho do genoma | Etiquetas SLAF recomendadas |
| <500MB | 100K ou WGS |
| 500 Mb- 1 Gb | 100 mil |
| 1 GB -2 GB | 200 mil |
| Genomas gigantes ou complexos | 300 - 400K |
| Formulários
| Recomendado Escala Populacional
| Estratégia e profundidade de sequenciamento
| |
| Profundidade
| Número da etiqueta
| ||
| GWAS
| Número da amostra ≥ 200
| 10X
|
De acordo com tamanho do genoma
|
| Evolução Genética
| Indivíduos de cada subgrupo ≥ 10; total de amostras ≥ 30
| 10X
| |
Entrega de amostra recomendada
Recipiente: tubo de centrífuga de 2 ml
Para a maioria das amostras, recomendamos não conservar em etanol.
Rotulagem das amostras: As amostras precisam ser claramente rotuladas e idênticas ao formulário de informações da amostra enviado.
Remessa: Gelo seco: As amostras precisam primeiro ser embaladas em sacos e enterradas em gelo seco.
Fluxo de trabalho de serviço






CQ de amostra
Experiência piloto
Experiência SLAF
Preparação da Biblioteca
Sequenciamento
Análise de dados
Serviços pós-venda
1. Estatísticas do resultado do mapa

2. Desenvolvimento de marcadores SLAF

3. Anotação de variação

| Ano | Diário | IF | Título | Formulários |
| 2022 | Comunicações da natureza | 17.694 | Base genômica dos giga-cromossomos e giga-genoma da peônia das árvores Paeonia ostii | SLAF-GWAS |
| 2015 | Novo Fitologista | 7.433 | Pegadas de domesticação ancoram regiões genômicas de importância agronômica em soja | SLAF-GWAS |
| 2022 | Jornal de Pesquisa Avançada | 12.822 | Introgressões artificiais de Gossypium barbadense em todo o genoma em G. hirsutum revelam loci superiores para melhoria simultânea da qualidade e rendimento da fibra de algodão características | SLAF-Genética evolutiva |
| 2019 | Planta Molecular | 10,81 | Análise genômica populacional e montagem De Novo revelam a origem de Weedy Arroz como um jogo evolutivo | SLAF-Genética evolutiva |
| 2019 | Genética da Natureza | 31.616 | Sequência do genoma e diversidade genética da carpa comum, Cyprinus carpio | Mapa de ligação SLAF |
| 2014 | Genética da Natureza | 25.455 | O genoma do amendoim cultivado fornece informações sobre cariótipos de leguminosas, poliplóides evolução e domesticação das culturas. | Mapa de ligação SLAF |
| 2022 | Revista de Biotecnologia Vegetal | 9.803 | A identificação de ST1 revela uma seleção envolvendo carona na morfologia da semente e teor de óleo durante a domesticação da soja | Desenvolvimento de marcador SLAF |
| 2022 | Revista Internacional de Ciências Moleculares | 6.208 | Identificação e desenvolvimento de marcadores de DNA para um mollis 2Ns de trigo-Leymus (2D) Substituição cromossômica disômica | Desenvolvimento de marcador SLAF |





