

Sequenciamento de mRNA sem referência-Illumina








Características
● Independente de qualquer genoma de referência,
● Os dados podem ser usados para analisar a estrutura e expressão das transcrições
● Identificar sites de recorte variáveis
Vantagens do serviço
● Entrega de resultados baseada em BMKCloud: Os resultados são entregues como arquivo de dados e relatório interativo por meio da plataforma BMKCloud, que permite uma leitura fácil de resultados de análises complexas e mineração de dados personalizada com base em análises de bioinformática padrão.
● Serviços pós-venda: Serviços pós-venda válidos por 3 meses após a conclusão do projeto, incluindo acompanhamento de projetos, solução de problemas, perguntas e respostas de resultados, etc.
Requisitos de amostra e entrega
Requisitos de amostra:
Nucleotídeos:
| Conc.(ng/μl) | Quantidade (μg) | Pureza | Integridade |
| ≥ 20 | ≥ 0,5 | DO260/280=1,7-2,5 DO260/230=0,5-2,5 Contaminação limitada ou inexistente de proteínas ou DNA mostrada no gel. | Para plantas: RIN≥6,5; Para animais: RIN≥7,0; 5,0≥28S/18S≥1,0; elevação limitada ou nenhuma elevação da linha de base |
Tecido: Peso (seco): ≥1g
*Para tecidos menores que 5 mg, recomendamos o envio de amostra de tecido congelado (em nitrogênio líquido).
Suspensão celular: Contagem de células = 3×107
*Recomendamos enviar lisado celular congelado.Caso a contagem de células seja menor que 5×105, recomenda-se o congelamento instantâneo em nitrogênio líquido.
Amostras de sangue:
PA×geneBloodRNATube;
6mLTRIzol e 2mL de sangue (TRIzol:Sangue=3:1)
Entrega de amostra recomendada
Recipiente:
Tubo de centrífuga de 2 ml (folha de estanho não é recomendada)
Rotulagem da amostra: Grupo+réplica, por exemplo, A1, A2, A3;B1, B2, B3... ...
Envio:
1.Gelo seco: As amostras precisam ser embaladas em sacos e enterradas em gelo seco.
2.Tubos RNAstable: As amostras de RNA podem ser secas em tubo de estabilização de RNA (por exemplo, RNAstable®) e enviadas em temperatura ambiente.
Fluxo de trabalho de serviço

Projeto de experimento

Entrega de amostra

Extração de RNA

Construção de biblioteca

Sequenciamento

Análise de dados

Serviços pós-venda
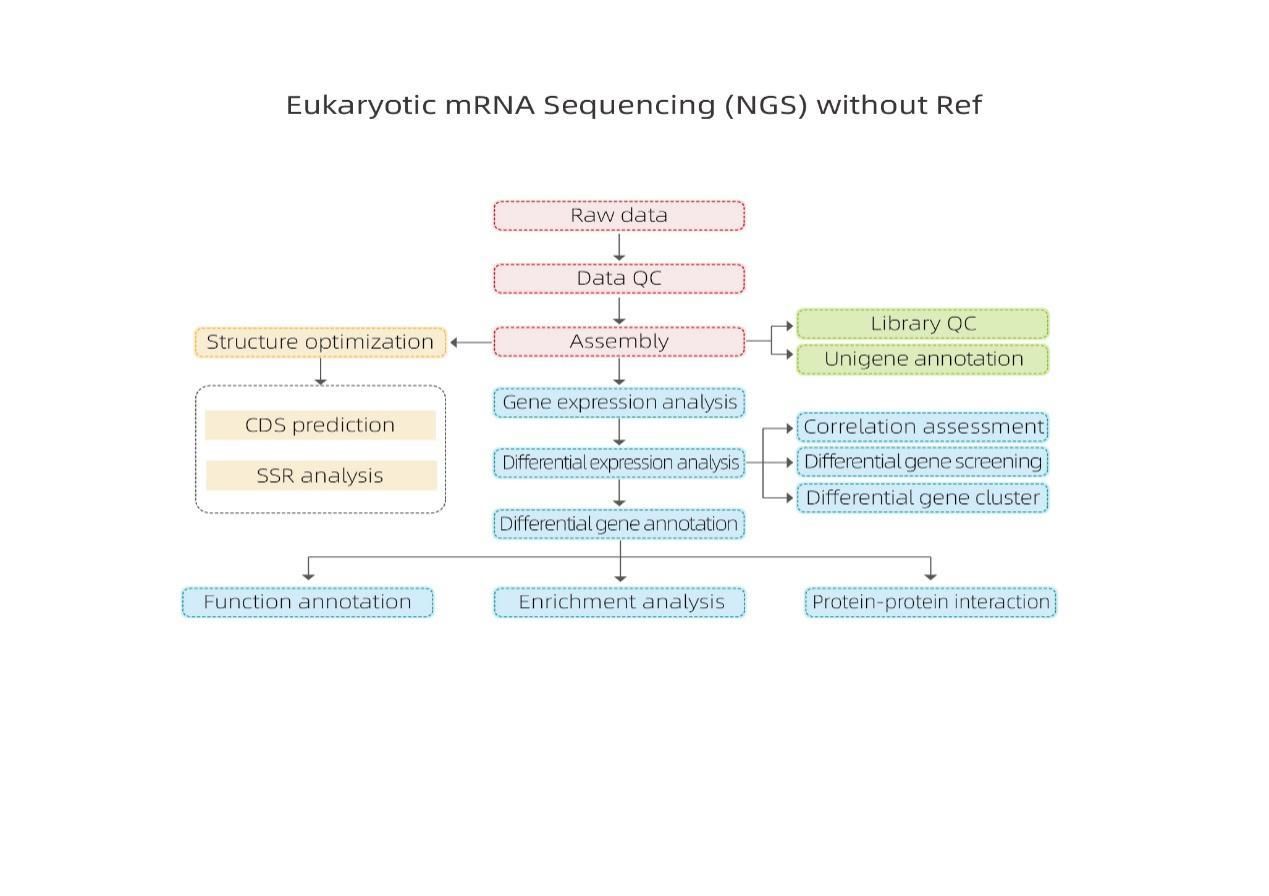
Bioinformática
1. Princípio de montagem do mRNA (denovo)
Pelo Trinity, as leituras são fragmentadas em pedaços menores, conhecidos como K-mer.Esses K-mers são então usados como sementes para serem estendidos em contigs e, em seguida, componentes baseados em sobreposições de contigs.Finalmente, De Bruijn foi aplicado aqui para reconhecer transcrições nos componentes.
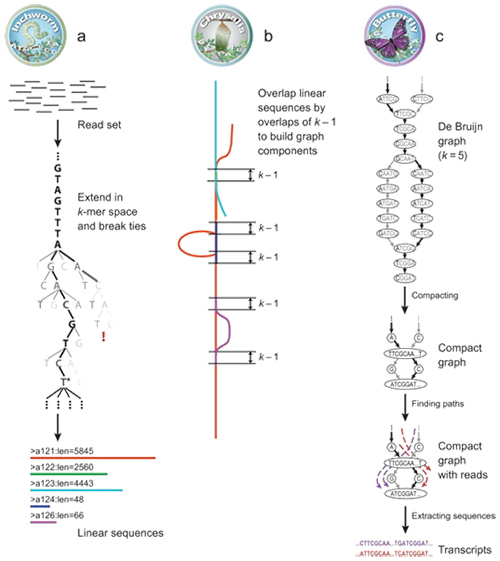
Visão geral do mRNA (de novo) do Trinity
2. Distribuição de mRNA (De novo) do nível de expressão gênica
O RNA-Seq é capaz de alcançar uma estimativa altamente sensível da expressão gênica.Normalmente, a faixa detectável de expressão de transcritos FPKM varia de 10 ^ -2 a 10 ^ 6
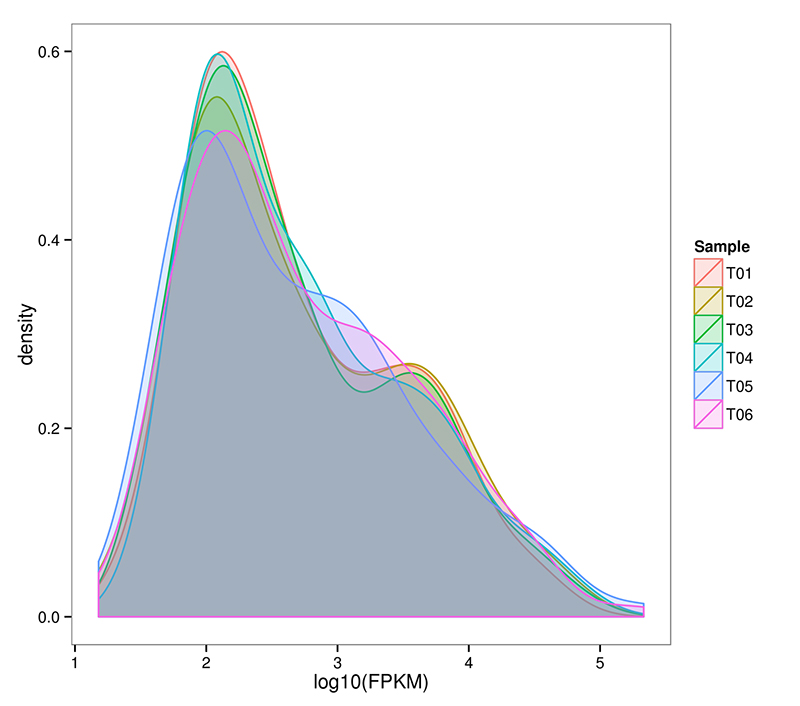
Distribuição de mRNA (De novo) da densidade de FPKM em cada amostra
3. Análise de Enriquecimento GO de mRNA (De novo) de DEGs
O banco de dados GO (Gene Ontology) é um sistema estruturado de anotação biológica que contém um vocabulário padrão de funções de genes e produtos genéticos.Ele contém vários níveis, onde quanto menor o nível, mais específicas são as funções.
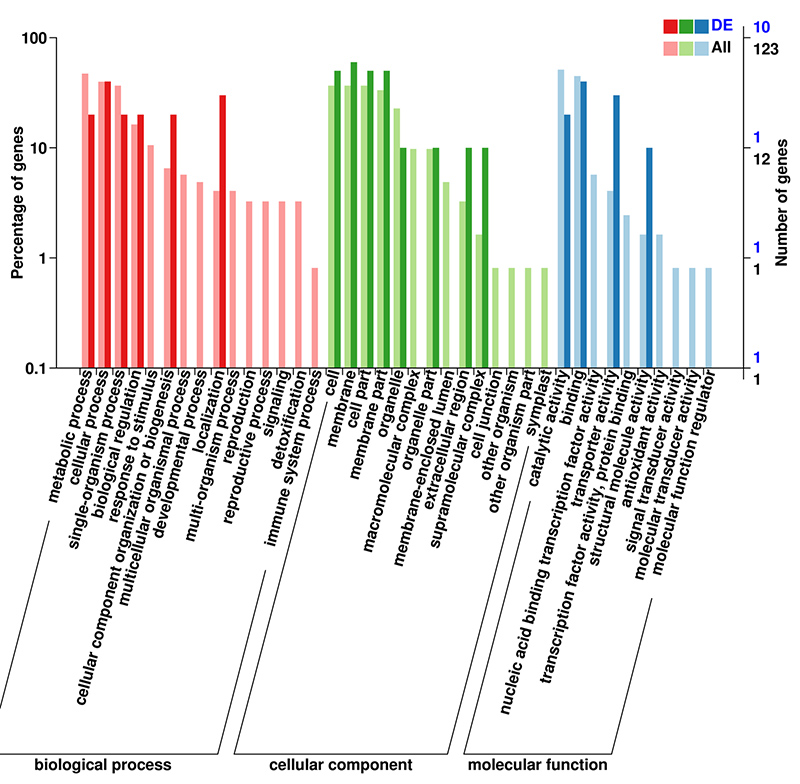
Classificação GO de mRNA (De novo) de DEGs no segundo nível
Caso BMK
Análise do transcriptoma do metabolismo da sacarose durante o inchaço e desenvolvimento do bulbo em cebola (Allium cepa L.)
Publicados: fronteiras na ciência das plantas,2016
Estratégia de sequenciamento
Illumina HiSeq2500
Coleta de amostras
A cultivar Utah Yellow Sweet Spain “Y1351” foi utilizada neste estudo.O número de amostras coletadas foi
15º dia após o inchaço (DAS) do bulbo (2 cm de diâmetro e 3–4 g de peso), 30º DAS (5 cm de diâmetro e 100–110 g de peso) e ∼3 no 40º DAS (7 cm de diâmetro e 260–300 gramas).
Principais resultados
1. no diagrama de Venn, um total de 146 DEGs foram detectados em todos os três pares de estágios de desenvolvimento
2. “Transporte e metabolismo de carboidratos” foi representado por apenas 585 unigenes (ou seja, 7% do COG anotado).
3. Os Unigenes anotados com sucesso no banco de dados GO foram classificados em três categorias principais para os três diferentes estágios de desenvolvimento do bulbo.Os mais representados na categoria principal “processo biológico” foram “processo metabólico”, seguido de “processo celular”.Na categoria principal “função molecular” as duas categorias mais representadas foram “ligação” e “atividade catalítica”.
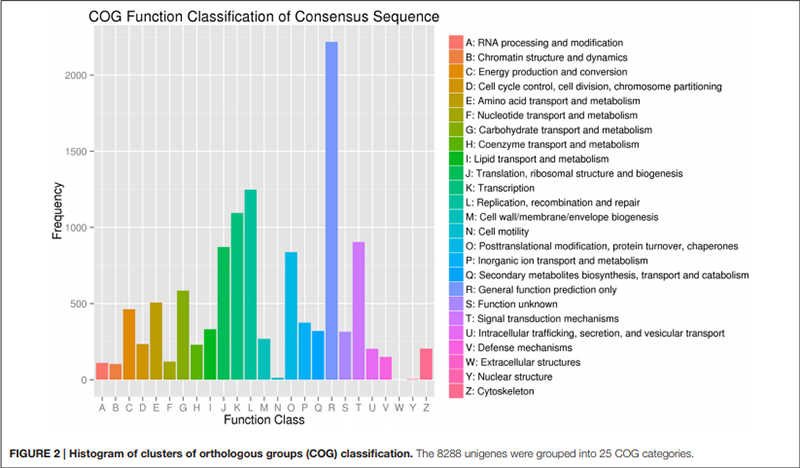 Histograma de classificação de clusters de grupos ortólogos (COG) | 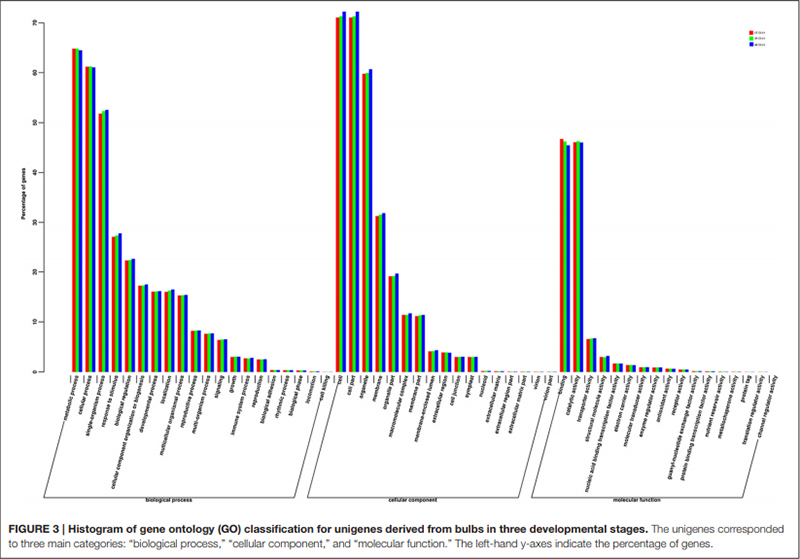 Histograma de classificação de ontologia genética (GO) para unigenes derivados de bulbos em três estágios de desenvolvimento |
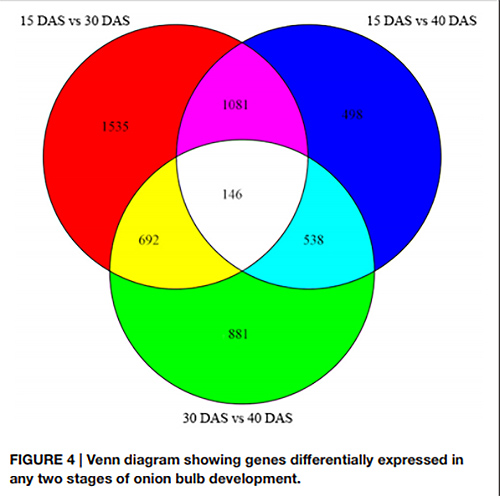 Diagrama de Venn mostrando genes expressos diferencialmente em quaisquer dois estágios de desenvolvimento do bulbo de cebola |
Referência
Zhang C, Zhang H, Zhan Z, et al.Análise do transcriptoma do metabolismo da sacarose durante o inchaço e desenvolvimento do bulbo em cebola (Allium cepa L.) [J].Fronteiras na Ciência das Plantas, 2016, 7:1425-.DOI: 10.3389/fpls.2016.01425





