

Genética Evolutiva








Vantagens do serviço
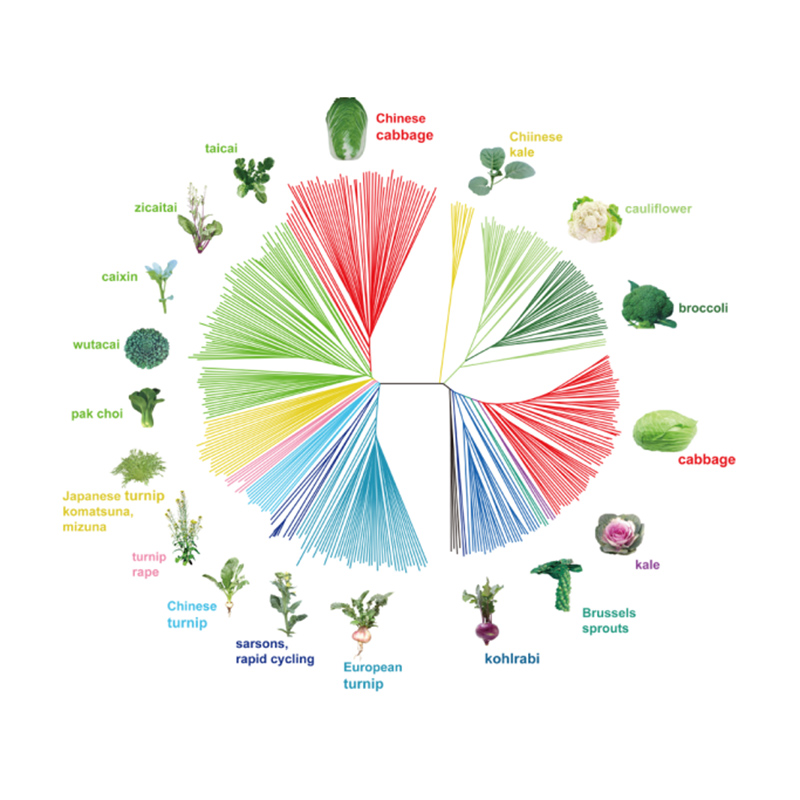
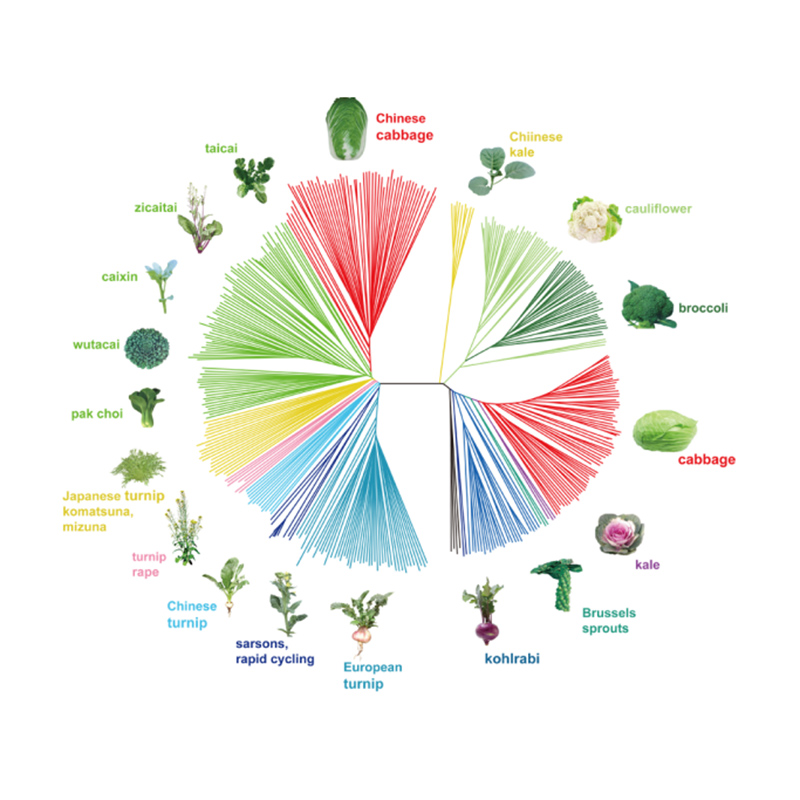
Takagi e outros,O diário da planta, 2013
● Estimar o tempo e a velocidade de divergência das espécies com base nas variações no nível de nucleotídeos e aminoácidos
● Revelação de relação filogenética mais confiável entre espécies com influência minimizada de evolução convergente e evolução paralela
● Construir ligações entre alterações genéticas e fenótipos para descobrir genes relacionados com características
● Estimativa da diversidade genética, que reflete o potencial evolutivo das espécies
● Tempo de resposta mais rápido
● Ampla experiência: A BMK acumulou enorme experiência em projetos populacionais e evolutivos por mais de 12 anos, cobrindo centenas de espécies, etc. e contribuiu em mais de 80 projetos de alto nível publicados em Nature Communications, Molecular Plants, Plant Biotechnology Journal, etc.
Especificações de serviço
Materiais:
Normalmente, são recomendadas pelo menos três subpopulações (por exemplo, subespécies ou estirpes).Cada subpopulação deve conter pelo menos 10 indivíduos (Plantas >15, podem ser reduzidas para espécies raras).
Estratégia de sequenciamento:
* O WGS pode ser empregado para espécies com genoma de referência de alta qualidade, enquanto o SLAF-Seq é aplicável a espécies com ou sem genoma de referência, ou genoma de referência de baixa qualidade.
| Aplicável ao tamanho do genoma | WGS | Tags SLAF (×10.000) |
| ≤ 500 MB | 10×/individual | WGS é mais recomendado |
| 500 MB - 1 GB | 10 | |
| 1 GB - 2 GB | 20 | |
| ≥2 GB | 30 |
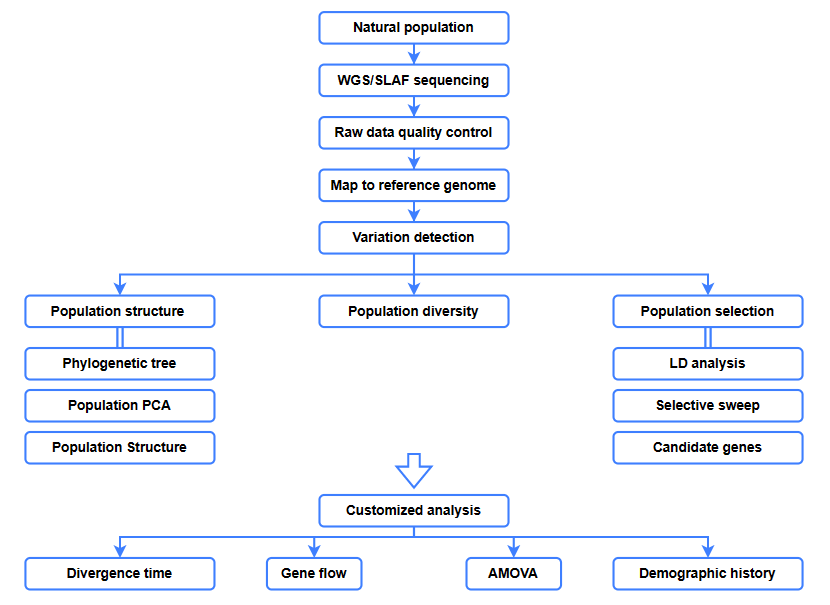
Análises de bioinformática
● Análise evolutiva
● Varredura seletiva
● Fluxo genético
● História demográfica
● Tempo de divergência
Requisitos de amostra e entrega
Requisitos de amostra:
| Espécies | Tecido | WGS-NGS | SLAF |
| Animal
| Tecido visceral |
0,5~1g
|
0,5g
|
| Tecido muscular | |||
| Sangue de mamífero | 1,5mL
| 1,5mL
| |
| Sangue de aves/peixe | |||
| Plantar
| Folha Fresca | 1~2g | 0,5~1g |
| Pétala/Caule | |||
| Raiz/Semente | |||
| Células | Célula cultivada |
| gDNA | Concentração | Quantia (eu) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1,6-2,5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Fluxo de trabalho de serviço

Projeto de experimento

Entrega de amostra

Construção de biblioteca

Sequenciamento

Análise de dados

Serviços pós-venda
*Os resultados de demonstração mostrados aqui são todos de genomas publicados com BMKGENE
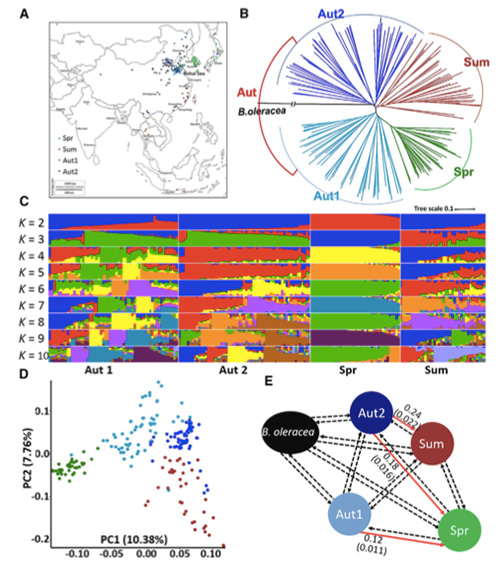
1. A análise da evolução contém a construção da árvore filogenética, estrutura populacional e PCA com base em variações genéticas.
A árvore filogenética representa relações taxonômicas e evolutivas entre espécies com ancestral comum.
O PCA visa visualizar a proximidade entre subpopulações.
A estrutura populacional mostra a presença de subpopulações geneticamente distintas em termos de frequências alélicas.
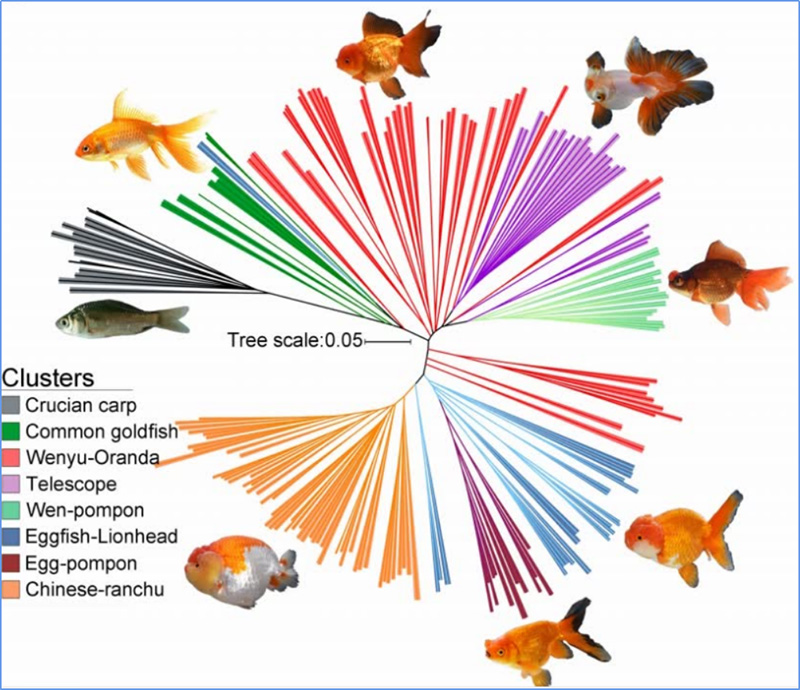
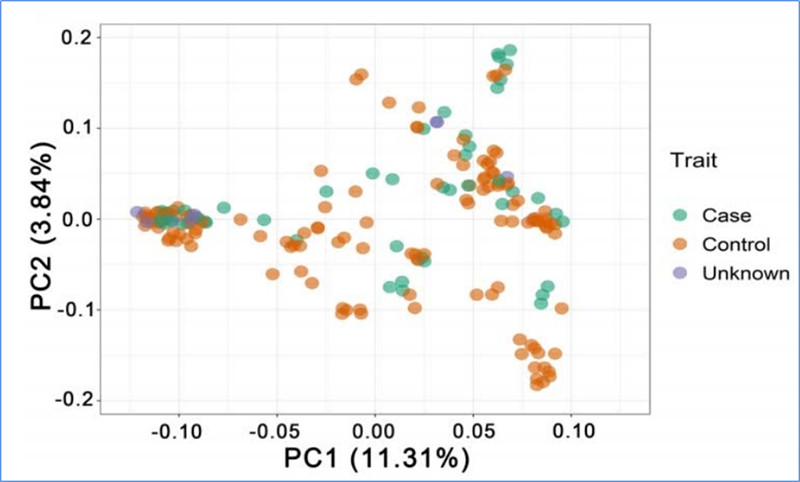
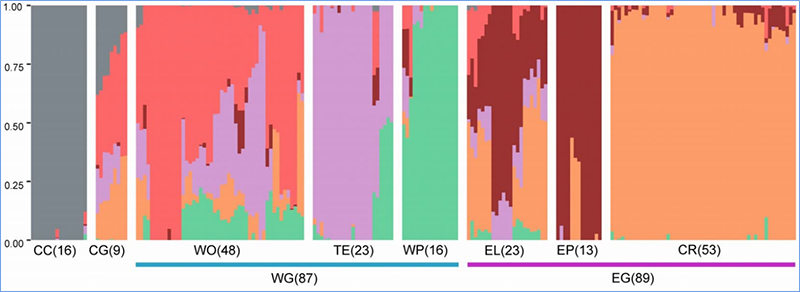
Chen, et.al.,PNAS, 2020
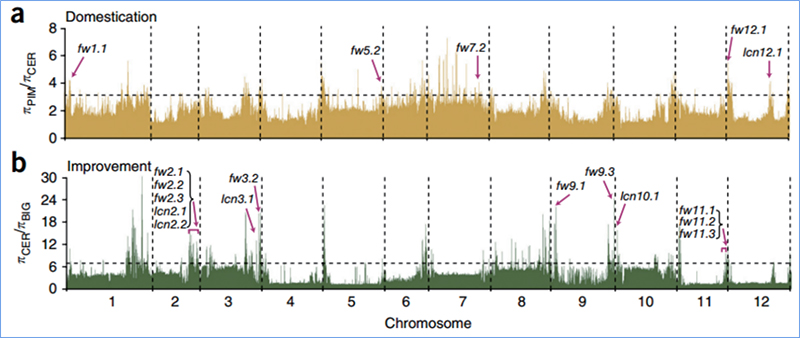
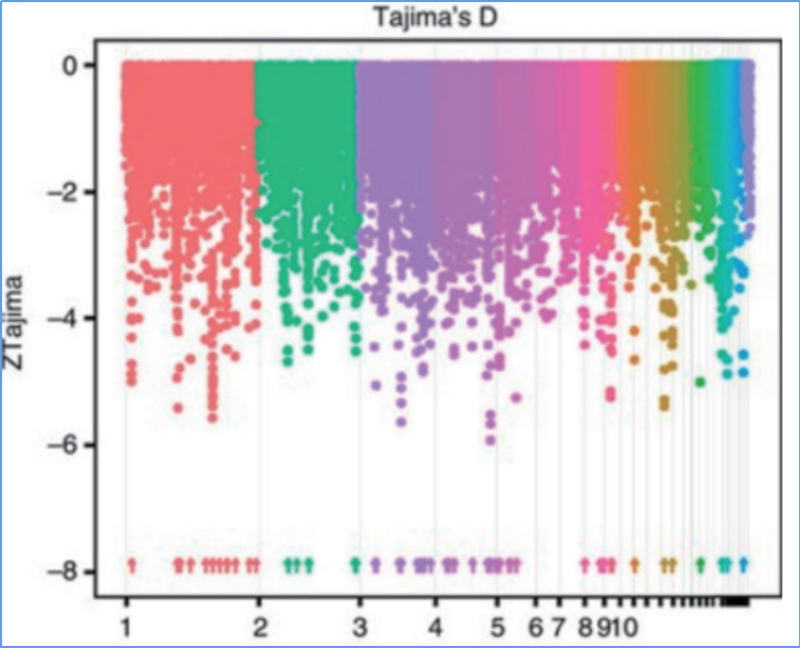
2. Varredura seletiva
A varredura seletiva refere-se a um processo pelo qual um local vantajoso é selecionado e as frequências de locais neutros vinculados são aumentadas e as de locais não vinculados são diminuídas, resultando na redução de regional.
A detecção de todo o genoma em regiões de varredura seletiva é processada calculando o índice genético populacional (π,Fst, D de Tajima) de todos os SNPs dentro de uma janela deslizante (100 Kb) em determinada etapa (10 Kb).
Diversidade de nucleotídeos (π)
D de Tajima
Índice de fixação (Fst)
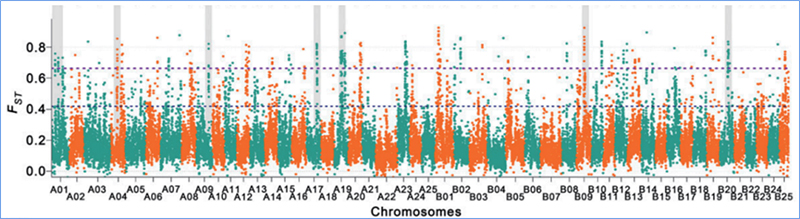
Wu, et.al.,Planta Molecular, 2018
3. Fluxo Genético
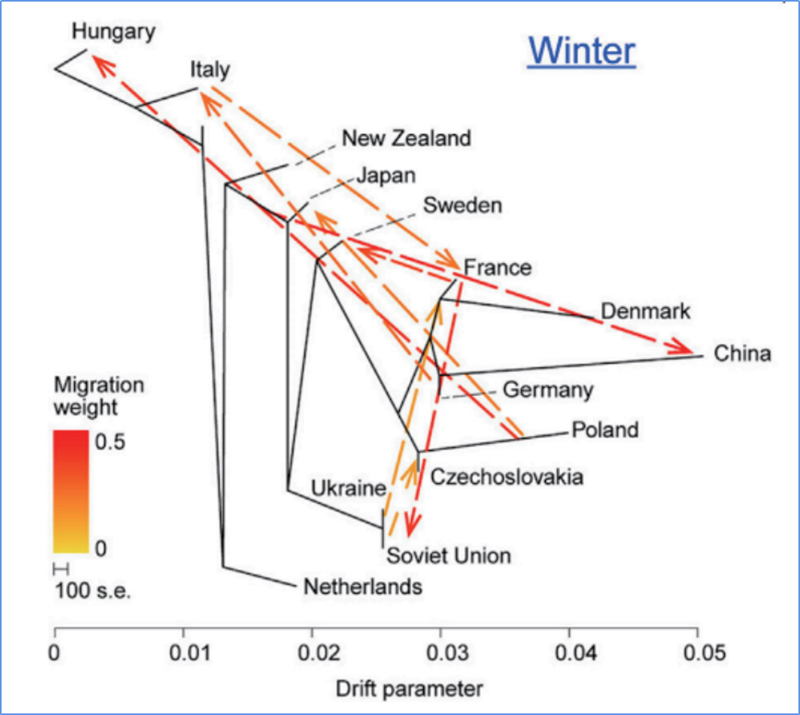
Wu, et.al.,Planta Molecular, 2018
4. História demográfica
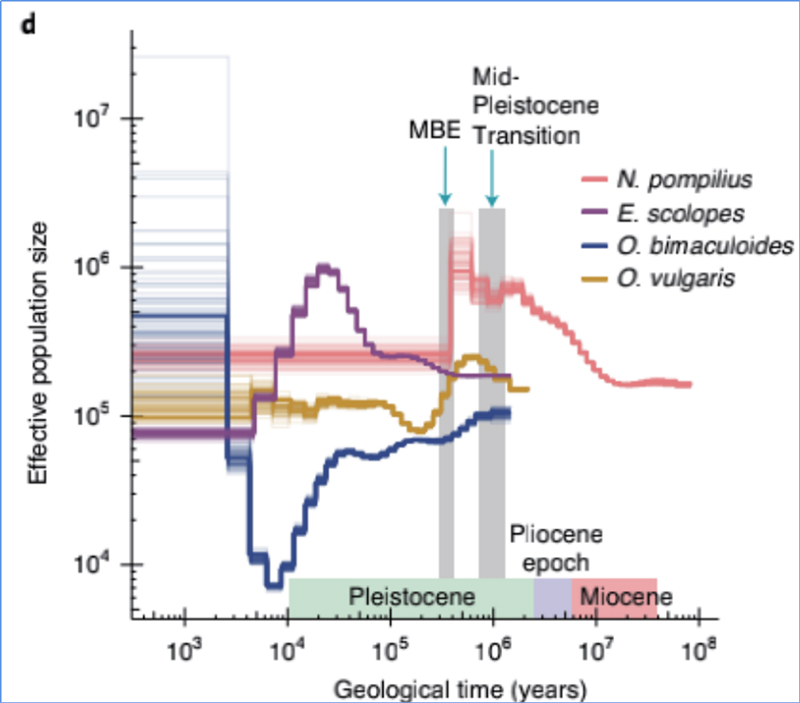
Zhang, et.al.,Ecologia e Evolução da Natureza, 2021
5. Tempo de divergência
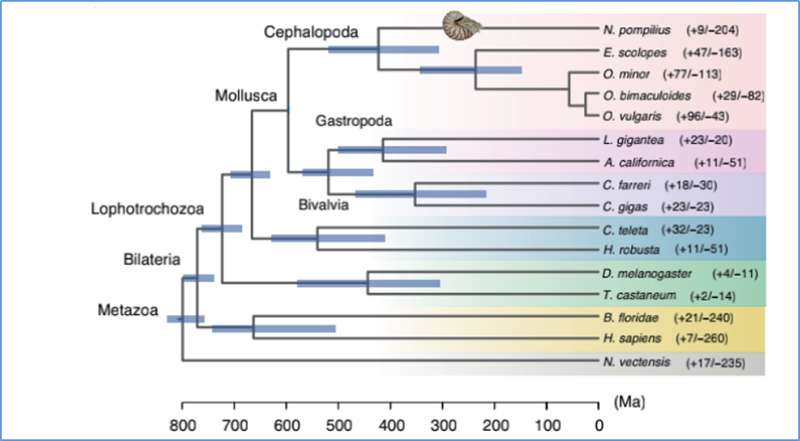
Zhang, et.al.,Ecologia e Evolução da Natureza, 2021
Caso BMK
Um mapa de variação genômica fornece insights sobre a base genética da seleção de couve chinesa de primavera (Brassica rapa ssp. Pekinensis)
Publicados: Planta Molecular, 2018
Estratégia de sequenciamento:
Resequenciamento: profundidade de sequenciamento: 10×
Principais resultados
Neste estudo, 194 couves chinesas foram processadas para re-sequenciamento com profundidade média de 10×, o que rendeu 1.208.499 SNPs e 416.070 InDels.A análise filogenética destas 194 linhagens mostrou que estas linhagens podem ser divididas em três ecótipos, primavera, verão e outono.Além disso, a estrutura populacional e a análise de PCA indicaram que o repolho chinês da primavera se originou de um repolho de outono em Shandong, China.Estas foram posteriormente introduzidas na Coreia e no Japão, cruzadas com linhas locais e algumas variedades de colheita tardia foram introduzidas na China e finalmente tornaram-se couve chinesa de primavera.
A varredura de todo o genoma em couves chinesas de primavera e couves de outono na seleção revelou 23 loci genômicos que passaram por forte seleção, dois dos quais foram sobrepostos à região de controle do tempo de aparafusamento com base no mapeamento de QTL.Descobriu-se que essas duas regiões contêm genes-chave que regulam o florescimento, BrVIN3.1 e BrFLC1.Esses dois genes foram ainda confirmados como envolvidos no tempo de aparafusamento por estudo do transcriptoma e experimentos transgênicos.
 Análise da estrutura populacional de couves chinesas | 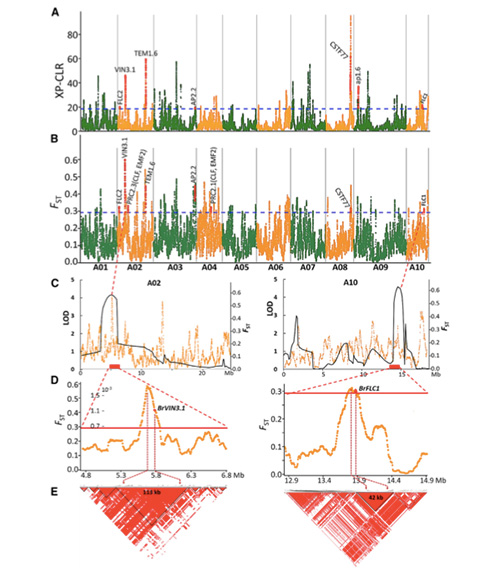 Informações genéticas na seleção de repolho chinês |
Tongbing, et al.“Um mapa de variação genômica fornece insights sobre a base genética da seleção de repolho chinês na primavera (Brassica rapa ssp.pekinensis).”Plantas Moleculares,11(2018):1360-1376.





