

Sequenciamento completo de mRNA -PacBio





Vantagens do serviço
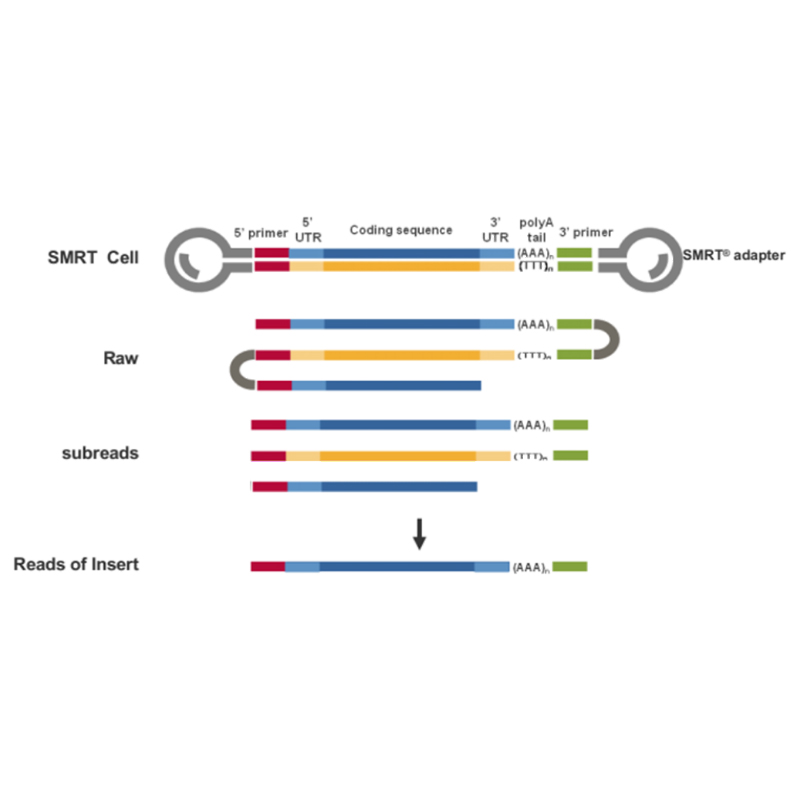
● Leitura direta da molécula de cDNA completa da extremidade 3' à extremidade 5'
● Resolução em nível de forma iso na estrutura de sequência
● Transcrições com alta precisão e integridade
● Altamente compatível com várias espécies
● Grande capacidade de sequenciamento com 4 plataformas de sequenciamento PacBio Sequel II equipadas
● Altamente experiente em mais de 700 projetos de sequenciamento de RNA baseados em Pacbio
● Entrega de resultados baseada em BMKCloud: mineração de dados personalizada disponível na plataforma.
● Serviços pós-venda válidos por 3 meses após a conclusão do projeto
Especificações de serviço
Plataforma: PacBio Sequel II
Biblioteca de sequenciamento: biblioteca de mRNA enriquecida com Poly A
Rendimento de dados recomendado: 20 Gb/amostra (dependendo da espécie)
FLNC(%): ≥75%
*FLNC: transcrições não quiméricas completas
Análises de bioinformática
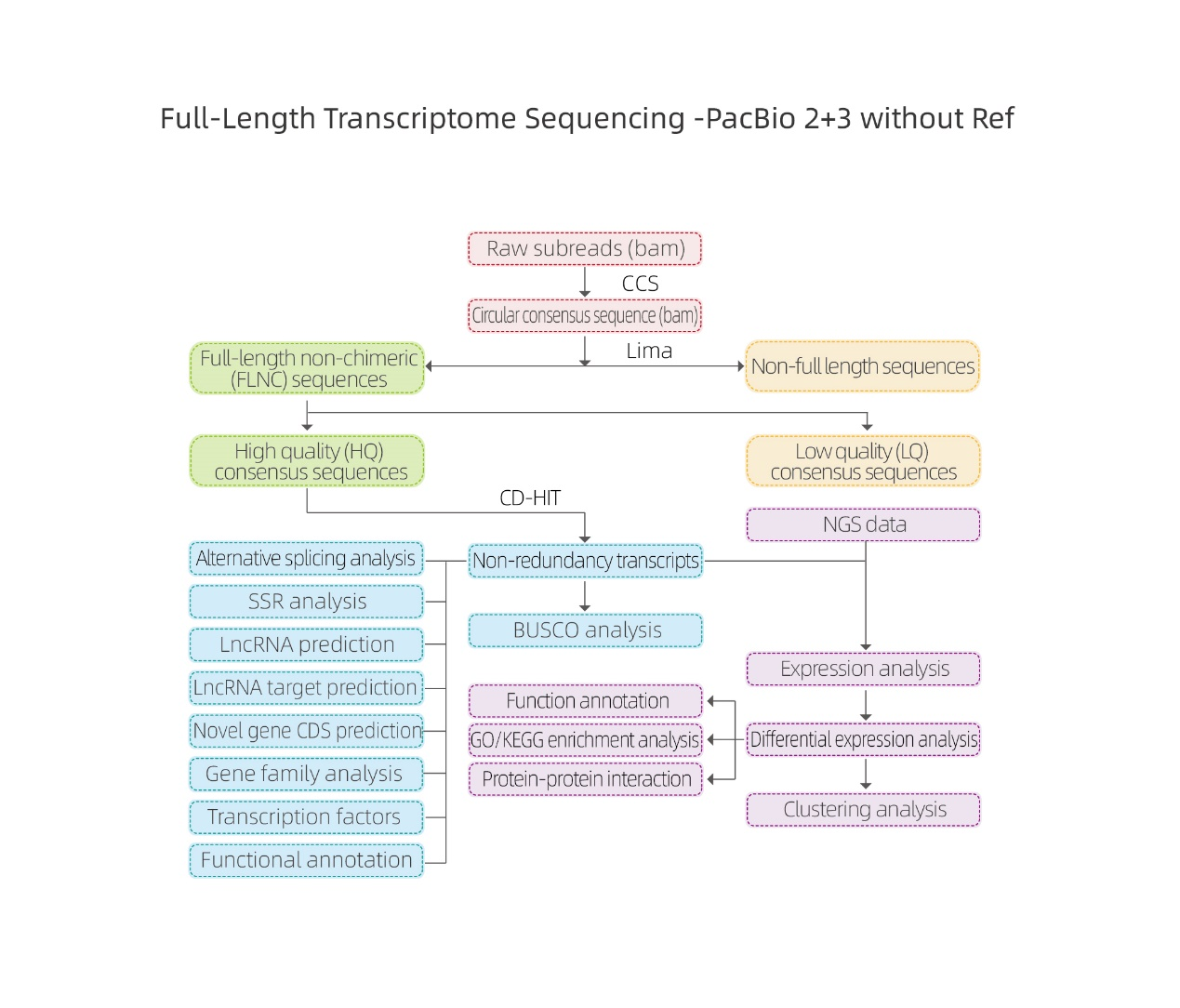
● Processamento de dados brutos
● Identificação da transcrição
● Estrutura de sequência
● Quantificação de Expressão
● Anotação de Função
Requisitos de amostra e entrega
Requisitos de amostra:
Nucleotídeos:
| Conc.(ng/μl) | Quantidade (μg) | Pureza | Integridade |
| ≥ 120 | ≥ 0,6 | DO260/280=1,7-2,5 DO260/230=0,5-2,5 Contaminação limitada ou inexistente de proteínas ou DNA mostrada no gel. | Para plantas: RIN≥7,5; Para animais: RIN≥8,0; 5,0≥ 28S/18S≥1,0; elevação limitada ou nenhuma elevação da linha de base |
Tecido: Peso (seco):≥1g
*Para tecidos menores que 5 mg, recomendamos o envio de amostra de tecido congelado (em nitrogênio líquido).
Suspensão celular:Contagem de células = 3×106- 1×107
*Recomendamos enviar lisado celular congelado.Caso a contagem de células seja menor que 5×105, recomenda-se o congelamento instantâneo em nitrogênio líquido, que é preferível para microextração.
Amostras de sangue:Volume≥1 mL
Microrganismo:Massa ≥ 1g
Entrega de amostra recomendada
Recipiente:
Tubo de centrífuga de 2 ml (folha de estanho não é recomendada)
Rotulagem da amostra: Grupo+réplica, por exemplo, A1, A2, A3;B1, B2, B3... ...
Envio:
1. Gelo seco: As amostras precisam ser embaladas em sacos e enterradas em gelo seco.
2. Tubos RNAstable: As amostras de RNA podem ser secas em tubo de estabilização de RNA (por exemplo, RNAstable®) e enviadas em temperatura ambiente.
Fluxo de trabalho de serviço

Projeto de experimento

Entrega de amostra

Extração de RNA

Construção de biblioteca

Sequenciamento

Análise de dados

Serviços pós-venda
1. Distribuição de comprimento FLNC
O comprimento da leitura não quimérica completa (FLNC) indica o comprimento do cDNA na construção da biblioteca.A distribuição de comprimento FLNC é um indicador crucial na avaliação da qualidade da construção de bibliotecas.
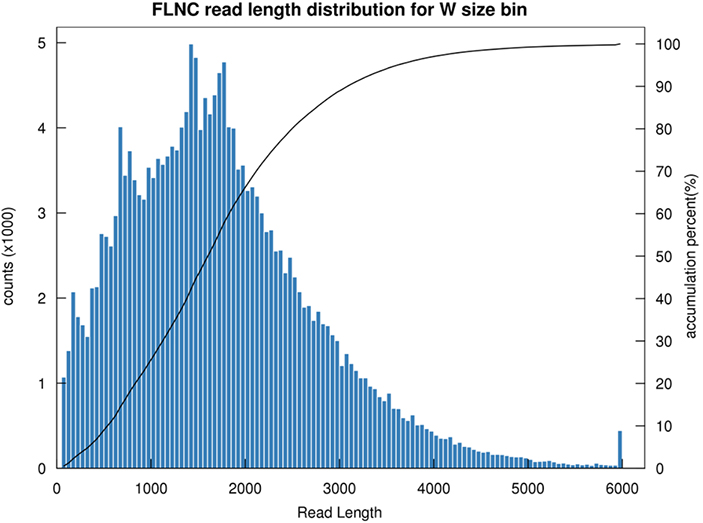
Distribuição de comprimento de leitura FLNC
2. Distribuição completa do comprimento da região ORF
Usamos o TransDecoder para prever regiões codificadoras de proteínas e sequências de aminoácidos correspondentes para gerar conjuntos unigenes, que contêm informações transcritas completas e não redundantes em todas as amostras.
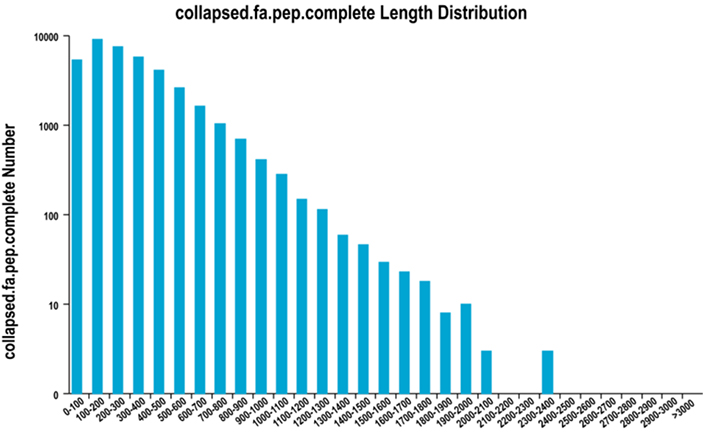
Distribuição completa do comprimento da região ORF
3. Análise de enriquecimento da via KEGG
Transcrições diferencialmente expressas (DETs) podem ser identificadas alinhando dados de sequenciamento de RNA baseados em NGS em conjuntos de transcrições completos gerados por dados de sequenciamento PacBio.Esses DETs podem ser processados posteriormente para diversas análises funcionais, por exemplo, análise de enriquecimento da via KEGG.
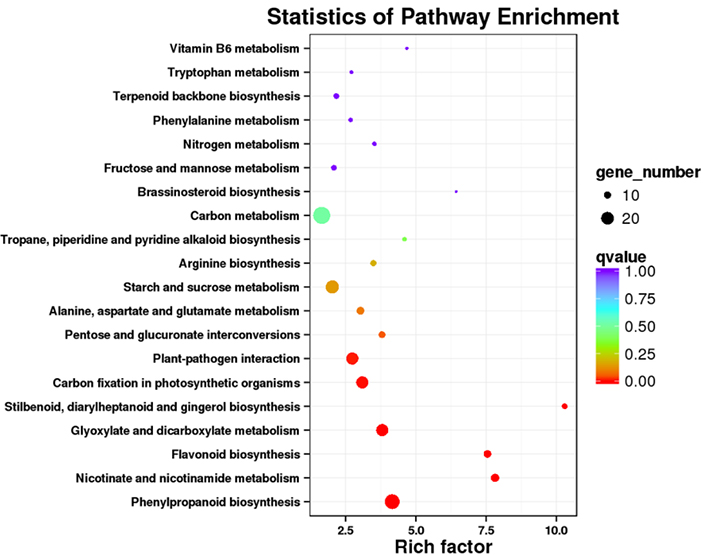
Enriquecimento da via DET KEGG - Gráfico de pontos
Caso BMK
A dinâmica de desenvolvimento do transcriptoma do caule de Populus
Publicados: Revista de Biotecnologia Vegetal, 2019
Estratégia de sequenciamento:
Coleta de amostras:regiões do caule: ápice, primeiro entrenó (IN1), segundo entrenó (IN2), terceiro entrenó (IN3), entrenó (IN4) e entrenó (IN5) de Nanlin895
Sequência NGS:RNA de 15 indivíduos foram reunidos como uma amostra biológica.Três réplicas biológicas de cada ponto foram processadas para a sequência NGS
Sequência TGS:As regiões do caule foram divididas em três regiões, ou seja, ápice, IN1-IN3 e IN4-IN5.Cada região foi processada para sequenciamento PacBio com quatro tipos de bibliotecas: 0-1 kb, 1-2 kb, 2-3 kb e 3-10 kb.
Principais resultados
1. Foram identificados um total de 87.150 transcrições completas, nas quais foram identificadas 2.081 novas isoformas e 62.058 novas isoformas de splicing alternativo.
Foram identificados 2.1187 lncRNA e 356 genes de fusão.
3. Do crescimento primário ao crescimento secundário, foram identificados 15.838 transcritos expressos diferencialmente de 995 genes expressos diferencialmente.Em todos os DEGs, 1.216 eram fatores de transcrição, a maioria dos quais ainda não foi relatada.
A análise de enriquecimento 4.GO revelou a importância da divisão celular e do processo de redução da oxidação no crescimento primário e secundário.
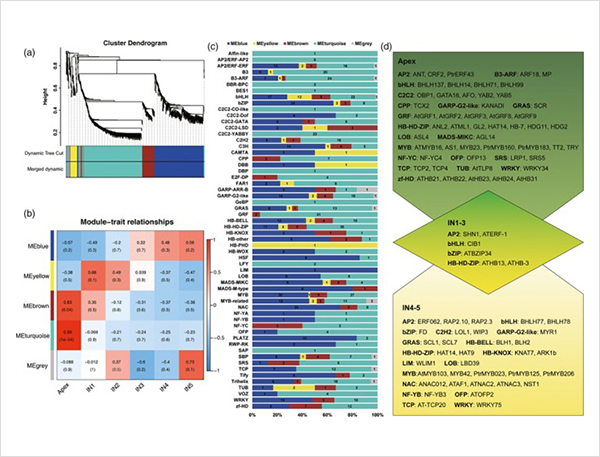
Eventos de splicing alternativos e diferentes isoformas
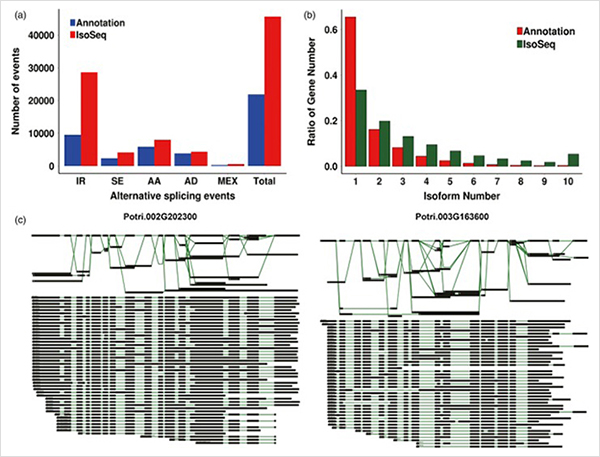
Análise WGCNA sobre fatores de transcrição
Referência
Chao Q, Gao ZF, Zhang D, et al.A dinâmica de desenvolvimento do transcriptoma do caule de Populus.Plant Biotechnol J. 2019;17(1):206-219.doi:10.1111/pbi.12958





