

Análise de segregante em massa






Vantagens do serviço
Takagi et al., The Plant Journal, 2013
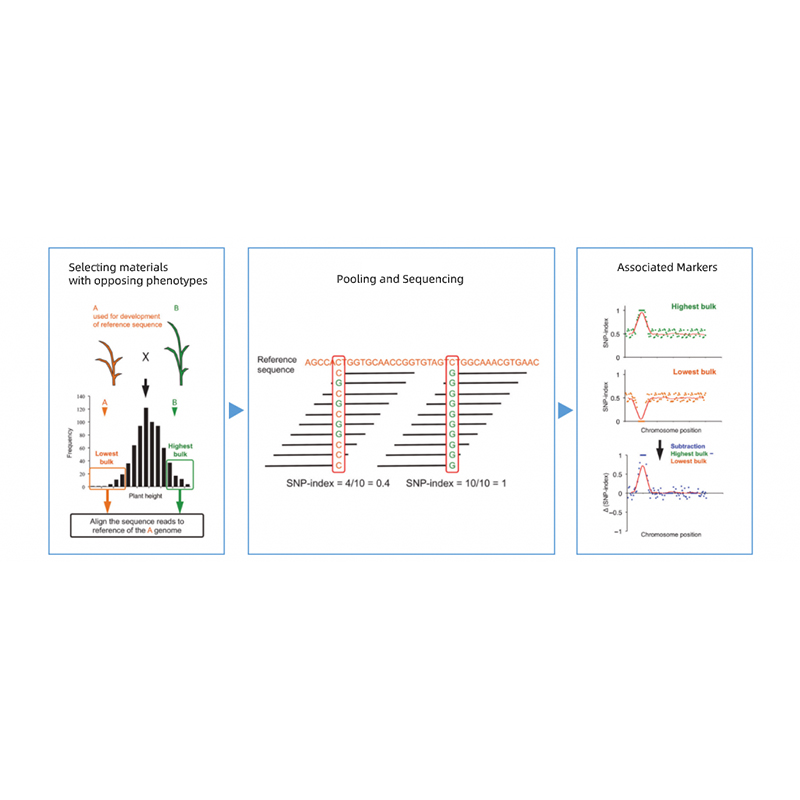
● Localização precisa: Misturar volumes com 30+30 a 200+200 indivíduos para minimizar ruído de fundo;previsão de região candidata baseada em mutantes não sinônimos.
● Análise abrangente: Anotação detalhada da função do gene candidato, incluindo NR, SwissProt, GO, KEGG, COG, KOG, etc.
● Tempo de resposta mais rápido: Localização rápida do gene em 45 dias úteis.
● Ampla experiência: a BMK contribuiu na localização de milhares de características, abrangendo diversas espécies como culturas, produtos aquáticos, florestas, flores, frutos, etc.
Especificações de serviço
População:
Segregação da descendência de pais com fenótipos opostos.
por exemplo, progênie F2, retrocruzamento (BC), linhagem endogâmica recombinante (RIL)
Piscina de mistura
Para características qualitativas: 30 a 50 indivíduos (mínimo 20)/volume
Para tratis quantitativos: 5% a 10% dos indivíduos com fenótipos extremos em toda a população (mínimo 30+30).
Profundidade de sequenciamento recomendada
Pelo menos 20X/indivíduo progenitor e 1X/filho (por exemplo, para um conjunto de mistura de descendentes de 30+30 indivíduos, a profundidade de sequenciamento será de 30X por grupo)
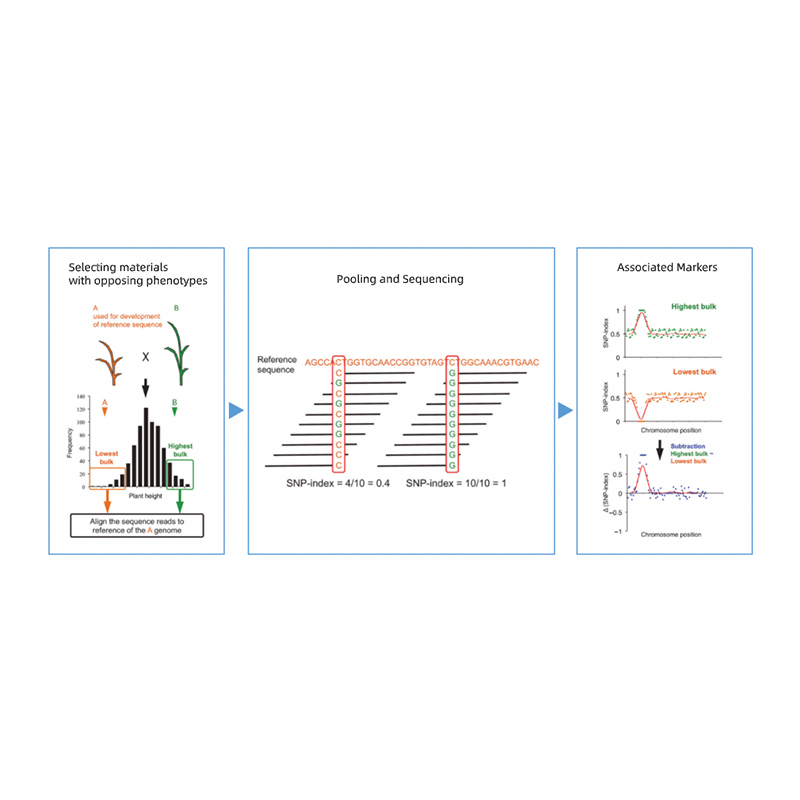
Análises de bioinformática
● Resequenciamento completo do genoma
● Processamento de dados
● Chamada SNP/Indel
● Triagem da região candidata
● Anotação da função do gene candidato
Requisitos de amostra e entrega
Requisitos de amostra:
Nucleotídeos:
| amostra de gDNA | Amostra de tecido |
| Concentração: ≥30 ng/μl | Plantas: 1-2g |
| Quantidade: ≥2 μg (Volume ≥15 μl) | Animais: 0,5-1g |
| Pureza: OD260/280 = 1,6-2,5 | Sangue total: 1,5 ml |
Fluxo de trabalho de serviço

Projeto de experimento

Entrega de amostra

Extração de RNA

Construção de biblioteca

Sequenciamento

Análise de dados

Serviços pós-venda
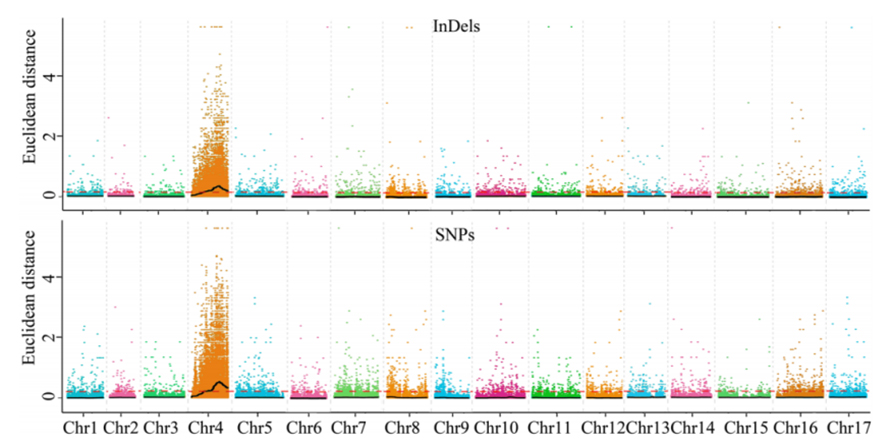
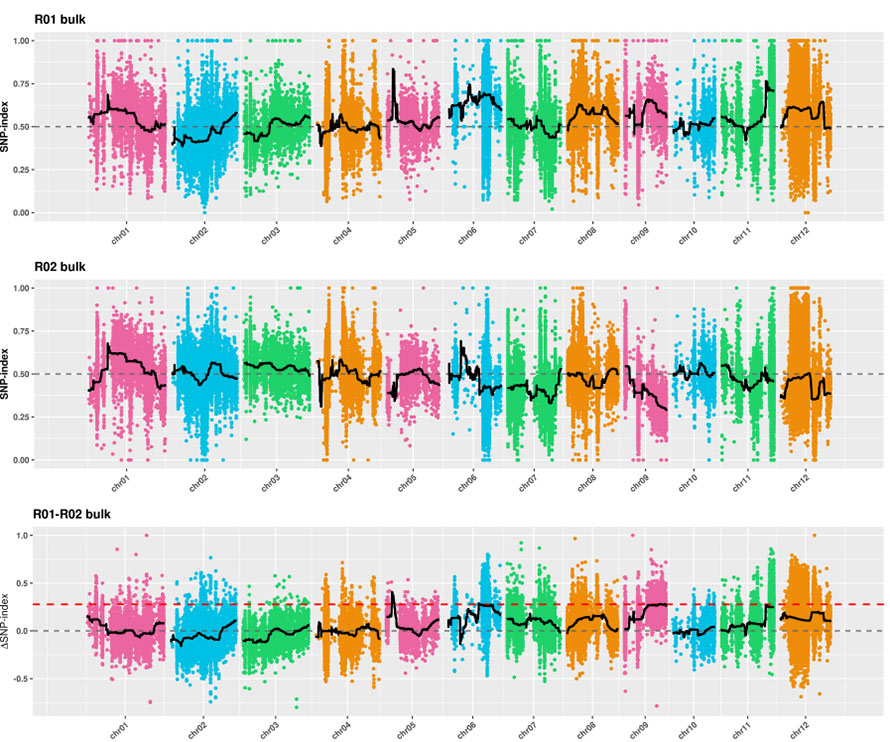
1. Análise de associação baseada na Distância Euclidiana (ED) para identificar a região candidata.Na figura a seguir
Eixo X: Número cromossômico;Cada ponto representa um valor ED de um SNP.A linha preta corresponde ao valor ED ajustado.Um valor ED mais alto indica uma associação mais significativa entre o local e o fenótipo.A linha tracejada vermelha representa o limite de associação significativa.
2. Análise de associação sem índice SNP
Eixo X: Número cromossômico;Cada ponto representa o valor do índice SNP.A linha preta representa o valor do índice SNP ajustado.Quanto maior o valor, mais significativa é a associação.
Caso BMK
O locus de característica quantitativa de efeito principal Fnl7.1 codifica uma proteína abundante em embriogênese tardia associada ao comprimento do pescoço do fruto em pepino
Publicados: Revista de Biotecnologia Vegetal, 2020
Estratégia de sequenciamento:
Pais (Jin5-508, YN): Resequenciamento do genoma completo para 34× e 20×.
Pools de DNA (50 de pescoço longo e 50 de pescoço curto): Resequenciamento para 61× e 52×
Principais resultados
Neste estudo, a população segregante (F2 e F2:3) foi gerada pelo cruzamento da linhagem de pepino de pescoço longo Jin5-508 e YN de pescoço curto.Dois pools de DNA foram construídos por 50 indivíduos com pescoço extremamente longo e 50 indivíduos com pescoço extremamente curto.O QTL de efeito principal foi identificado no Chr07 pela análise BSA e mapeamento tradicional de QTL.A região candidata foi ainda mais reduzida por mapeamento fino, quantificação da expressão gênica e experimentos transgênicos, que revelaram o gene chave no controle do comprimento do pescoço, CsFnl7.1.Além disso, descobriu-se que o polimorfismo na região promotora CsFnl7.1 estava associado à expressão correspondente.Análises filogenéticas adicionais sugeriram que o locus Fnl7.1 é muito provavelmente originado da Índia.
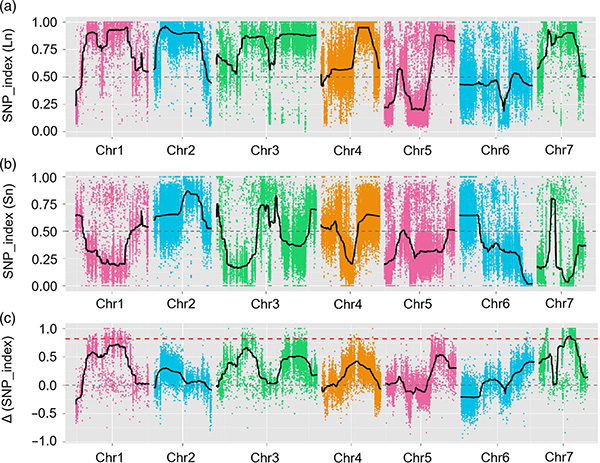 Mapeamento de QTL na análise de BSA para identificar região candidata associada ao comprimento do pescoço do pepino | 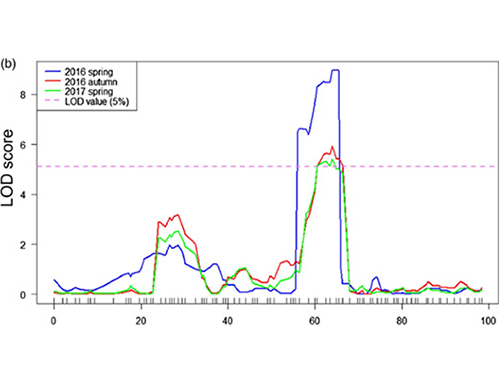 Perfis LOD de QTL de comprimento de pescoço de pepino identificados em Chr07 |
Xu, X., et al.“O locus de característica quantitativa de efeito principal Fnl7.1 codifica uma proteína abundante em embriogênese tardia associada ao comprimento do pescoço do fruto no pepino.”Jornal de Biotecnologia Vegetal 18.7(2020).





