-

Chromatin Immunoprecipitation Sequencing (ChIP-seq)
Chromatin Immunoprecipitation (CHIP) is a technique that leverages antibodies to selectively enrich DNA-binding proteins and their corresponding genomics targets. Its integration with NGS enables the genome-wide profiling of DNA targets associated with histone modification, transcription factors, and other DNA-binding proteins. This dynamic approach enables comparisons of binding sites across diverse cell types, tissues, or conditions. ChIP-Seq’s applications span from studying transcriptional regulation and developmental pathways to elucidating disease mechanisms, making it an indispensable tool for understanding genomic regulation landscapes and advancing therapeutic insights.
Platform: Illumina NovaSeq
-

Metagenomic Sequencing -NGS

A metagenome is a collection of the total genetic material of a mixed community of organisms, such as environmental and human metagenomes. It contains genomes of both cultivatable and uncultivatable microorganisms. Shotgun metagenomic sequencing with NGS enables the study of these intricate genomic landscapes embedded in environmental samples by providing more than taxonomic profiling, giving also granular insights into species diversity, abundance dynamics, and complex population structures. Beyond taxonomic studies, shotgun metagenomics also offers a functional genomics perspective, enabling the exploration of encoded genes and their putative roles in ecological processes. Finally, the establishment of correlation networks between genetic elements and environmental factors contributes to a holistic understanding of the intricate interplay between microbial communities and their ecological background. In conclusion, metagenomic sequencing stands as a pivotal instrument for unravelling the genomic intricacies of diverse microbial communities, illuminating the multifaceted relationships between genetics and ecology within these complex ecosystems.
Platforms: Illumina NovaSeq and DNBSeq T7
-

Metagenomic Sequencing-Nanopore

A metagenome is a collection of the genetic material of a mixed community of organisms, such as environmental and human metagenomes. It contains genomes of both cultivatable and uncultivatable microorganisms. Metagenomic sequencing enables the study of these intricate genomic landscapes embedded in ecological samples by providing more than taxonomic profiling. It also offering a functional genomics perspective by exploring the encoded genes and their putative roles in environmental processes. While traditional shotgun approaches with Illumina sequencing have been widely used in metagenomic studies, the advent of Nanopore long-read sequencing has changed the field. Nanopore technology enhances downstream bioinformatic analyses, notably metagenome assembly, ensuring more continuous assemblies. Reports indicate that Nanopore-based metagenomics has successfully generated complete and closed bacterial genomes from complex microbiomes (Moss, E. L., et al., Nature Biotech, 2020). Integrating Nanopore reads with Illumina reads provides a strategic approach for error correction, mitigating Nanopore’s inherent low accuracy. This synergistic combination leverages the strengths of each sequencing platform, offering a robust solution to overcome potential limitations and advancing the precision and reliability of metagenomic analyses.
-

Whole genome bisulfite sequencing(WGBS)
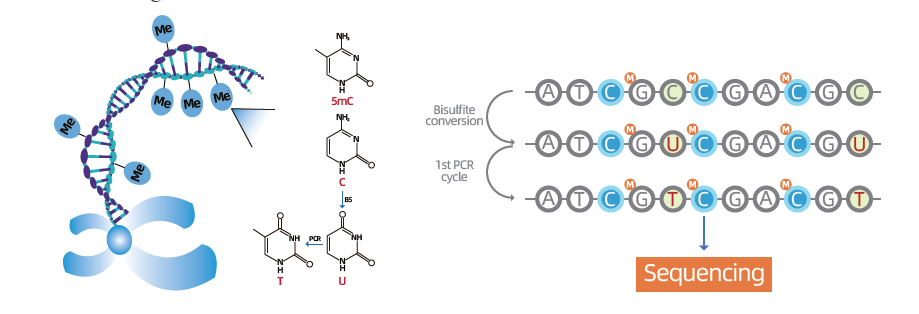
Whole Genome Bisulfite Sequencing (WGBS) stands as the gold-standard methodology for in-depth exploration of DNA methylation, specifically the fifth position in cytosine (5-mC), a pivotal regulator of gene expression and cellular activity. The principle underlying WGBS involves bisulfite treatment, inducing the conversion of unmethylated cytosines to uracil (C to U), while leaving methylated cytosines unchanged. This technique offers single-base resolution, allowing researchers to comprehensively investigate the methylome and uncover abnormal methylation patterns associated with various conditions, notably cancer. By employing WGBS, scientists can gain unparalleled insights into genome-wide methylation landscapes, providing a nuanced understanding of the epigenetic mechanisms that underlie diverse biological processes and diseases.
-

Assay for Transposase-Accessible Chromatin with High Throughput Sequencing (ATAC-seq)
ATAC-seq is a high-throughput sequencing technique used for genome-wide chromatin accessibility analysis. It use provides deeper understanding of the complex mechanisms of global epigenetic control over gene expression. The method uses a hyperactive Tn5 transposase to simultaneously fragment and tag open chromatin regions by inserting sequencing adaptors. Subsequent PCR amplification results in the creation of a sequencing library, which allows for the comprehensive identification of open chromatin regions under specific space-time conditions. ATAC-seq provides a holistic view of accessible chromatin landscapes, unlike methods that solely focus on transcription factor binding sites or specific histone-modified regions. By sequencing these open chromatin regions, ATAC-seq reveals regions more likely to active regulatory sequences and potential transcription factor binding sites, offering valuable insights into the dynamic modulation of gene expression across the genome.
-

16S/18S/ITS Amplicon Sequencing-PacBio
The 16S and 18S rRNA genes, along with the Internal Transcribed Spacer (ITS) region, serve as pivotal molecular fingerprinting markers due to their combination of highly conserved and hyper-variable regions, making them invaluable tools for characterizing prokaryotic and eukaryotic organisms. Amplification and sequencing of these regions offer an isolation-free approach for investigating the microbial composition and diversity across various ecosystems. While Illumina sequencing typically targets short hypervariable regions like V3-V4 of 16S and ITS1, it has been demonstrated that superior taxonomic annotation is achievable by sequencing the full length of 16S, 18S, and ITS. This comprehensive approach results in higher percentages of accurately classified sequences, achieving a level of resolution that extends to species identification. PacBio’s Single-Molecule Real-Time (SMRT) sequencing platform stands out by providing highly accurate long reads (HiFi) that cover the full-length amplicons, rivalling the precision of Illumina sequencing. This capability allows researchers to attain an unmatched advantage — a panoramic view of the genetic landscape. The extended coverage significantly elevates the resolution in species annotation, particularly within bacterial or fungal communities, enabling a deeper understanding of the intricacies of microbial populations.
-

16S/18S/ITS Amplicon Sequencing-NGS
Amplicon sequencing with Illumina technology, specifically targeting the 16S, 18S, and ITS genetic markers, is a powerful method for unraveling the phylogeny, taxonomy, and species abundance within microbial communities. This approach involves sequencing the hypervariable regions of housekeeping genetic markers. Originally introduced as a molecular fingerprint by Woeses et al in 1977, this technique has revolutionized microbiome profiling by enabling isolation-free analyses. Through the sequencing of 16S (bacteria), 18S (fungi), and Internal Transcribed Spacer (ITS, fungi), researchers can identify not only abundant species but also rare and unidentified ones. Widely adopted as a pivotal tool, amplicon sequencing has become instrumental in discerning differential microbial compositions across diverse environments, including the human mouth, intestines, stool, and beyond.
-

Bacterial and Fungal Whole Genome Re-Sequencing

Bacterial and fungal whole-genome re-sequencing projects are pivotal for advancing microbial genomics by enabling the completion and comparison of microbial genomes. This facilitates fermentation engineering, the optimization of industrial processes, and the exploration of secondary metabolism pathways. Furthermore, fungal and bacterial re-sequencing is crucial for understanding environmental adaptation, optimizing strains, and revealing genetic evolution dynamics, with broad implications in medicine, agriculture, and environmental science.
-

PacBio-Full-length 16S/18S/ITS Amplicon Sequencing
Amplicon (16S/18S/ITS) platform is developed with years of experience in microbial diversity project analysis, which contains standardized basic analysis and personalized analysis: basic analysis covers the mainstream analysis content of current microbial research, the analysis content is rich and comprehensive, and analysis results are presented in the form of project reports; The content of personalized analysis is diverse. Samples can be selected and parameters can be set flexibly according to the basic analysis report and research purpose, to realize personalized requirements. Windows operating system, simple and fast.
-

PacBio-Full-length Transcriptome (Non-Reference)
Taking Pacific Biosciences (PacBio) Isoform sequencing data as input, this App is able to identify full-length transcript sequences (without assembly). By mapping full-length sequences against reference genome, transcripts can be optimized by known genes, transcripts, coding regions, etc. In this case, more accurate identification of mRNA structures, such as alternative splicing, etc, can be achieved. Joint analysis with NGS transcriptome sequencing data enables more comprehensive annotation and more accurate quantification in expression at transcript level, which largely benefits downstream differential expression and functional analysis.
-

Reduced Representation Bisulfite Sequencing (RRBS)

Reduced Representation Bisulfite Sequencing (RRBS) has emerged as a cost-effective and efficient alternative to Whole Genome Bisulfite Sequencing (WGBS) in DNA methylation research. While WGBS provides comprehensive insights by examining the entire genome at single base resolution, its high cost can be a limiting factor. RRBS strategically mitigates this challenge by selectively analyzing a representative portion of the genome. This methodology relies on the enrichment of CpG island-rich regions by MspI cleavage followed by size selection of 200-500/600 bps fragments. Consequently, only regions proximal to CpG islands are sequenced, while those with distant CpG islands are excluded from the analysis. This process, combined with bisulfite sequencing, allows for high-resolution detection of DNA methylation, and the sequencing approach, PE150, focuses specifically on the ends of the inserts rather than the middle, increasing the efficiency of methylation profiling. The RRBS is an invaluable tool that enables cost-effective DNA methylation research and advances knowledge of epigenetic mechanisms.
-

Prokaryotic mRNA Sequencing
mRNA sequencing empowers the comprehensive profiling of all mRNA transcripts within cells under specific conditions. This cutting-edge technology serves as a potent tool, unveiling intricate gene expression profiles, gene structures, and molecular mechanisms associated with diverse biological processes. Widely adopted in fundamental research, clinical diagnostics, and drug development, mRNA sequencing offers insights into the intricacies of cellular dynamics and genetic regulation. Our prokaryotic mRNA sample processing is tailored for prokaryotic transcriptomes, involving rRNA depletion and directional library preparation.
Platform: Illumina NovaSeq X
-

Metatranscriptome Sequencing
Leveraging Illumina sequencing technology, BMKGENE’s metatranscriptome sequencing service unveils the dynamic gene expression of a diverse array of microbes, spanning eukaryotes to prokaryotes and viruses, within natural environments such as soil, water, sea, stool, and the gut. Our comprehensive service empowers researchers to delve into complex microbial communities’ complete gene expression profiles. Beyond taxonomic analysis, our metatranscriptome sequencing service facilitates exploration into functional enrichment, shedding light on differentially expressed genes and their roles. Uncover a wealth of biological insights as you navigate the complex landscapes of gene expression, taxonomic diversity, and functional dynamics within these diverse environmental niches.