

Sekwencjonowanie fragmentów amplifikowanych w specyficznym locus (SLAF-Seq)





Szczegóły usługi
Schemat techniczny
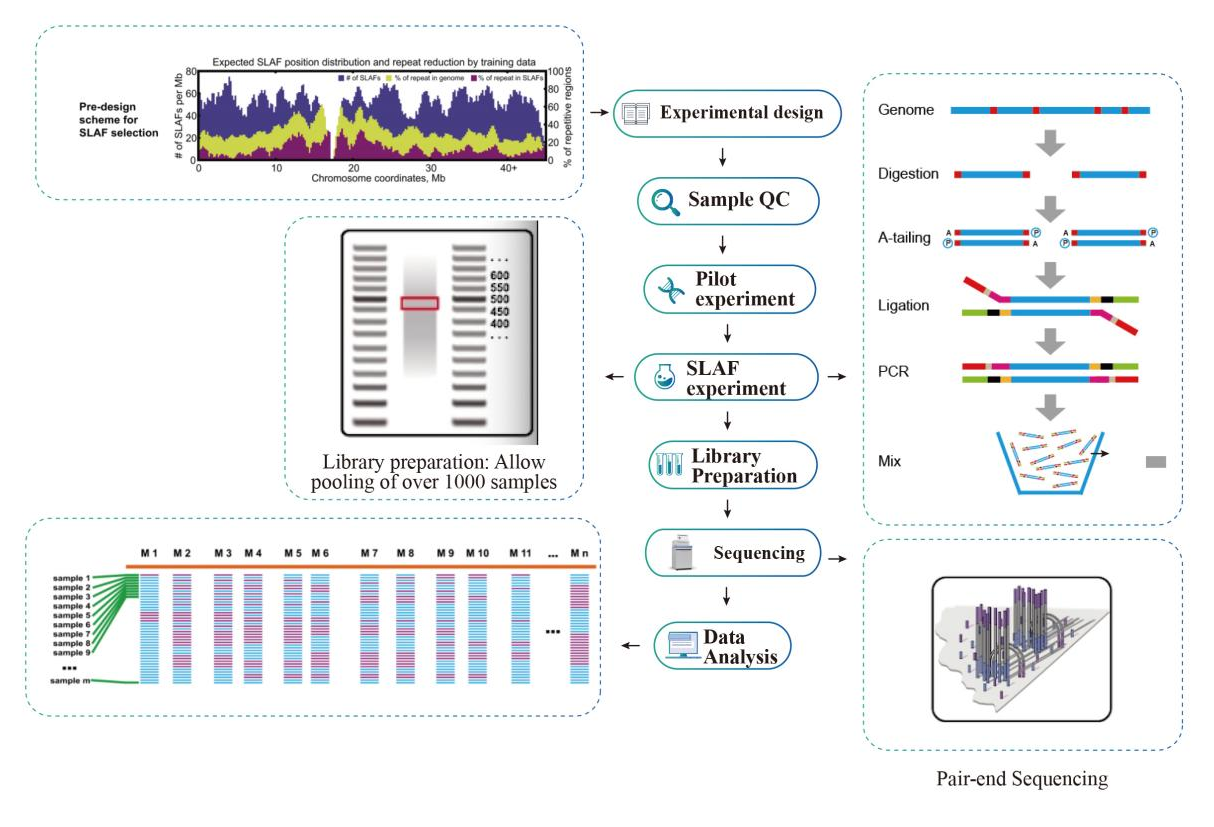
Przepływ pracy

Zalety serwisu
Wysoka skuteczność wykrywania markerów- Wysokoprzepustowa technologia sekwencjonowania pomaga SLAF-Seq w odkrywaniu setek tysięcy znaczników w całym genomie.
Niska zależność od genomu- Można go zastosować do gatunków z genomem referencyjnym lub bez niego.
Elastyczny projekt schematu- Trawienie jednoenzymatyczne, dwuenzymatyczne, wieloenzymatyczne i różne typy enzymów, wszystkie można wybrać w celu zaspokojenia różnych celów badawczych lub gatunków.Aby zapewnić optymalny projekt enzymu, stosuje się ocenę wstępną in silico.
Efektywne trawienie enzymatyczne- Przeprowadzono eksperyment wstępny w celu optymalizacji warunków, dzięki czemu eksperyment formalny jest stabilny i niezawodny.Wydajność zbierania fragmentów może osiągnąć ponad 95%.
Równomiernie rozmieszczone znaczniki SLAF- Znaczniki SLAF są w największym stopniu równomiernie rozmieszczone we wszystkich chromosomach, osiągając średnio 1 SLAF na 4 kb.
Skuteczne unikanie powtórzeń- Sekwencja powtórzeń w danych SLAF-Seq jest zmniejszona do wartości poniżej 5%, szczególnie u gatunków o wysokim poziomie powtórzeń, takich jak pszenica, kukurydza itp.
Duże doświadczenie-Ponad 2000 zamkniętych projektów SLAF-Seq dotyczących setek gatunków obejmujących rośliny, ssaki, ptaki, owady, organizmy wodne itp.
Opracowany samodzielnie bioinformatyczny przepływ pracy- W BMKGENE opracowano zintegrowany bioinformatyczny przepływ pracy dla SLAF-Seq, aby zapewnić niezawodność i dokładność wyników końcowych.
Specyfikacje usług
| Platforma | Stężenie (ng/gl) | Całkowity (ug) | OD260/280 |
| Illumina NovaSekw | >35 | >1.6(Objętość> 15μl) | 1,6-2,5 |
Zalecana strategia sekwencjonowania
Głębokość sekwencjonowania: 10X/tag
| Rozmiar genomu | Polecane tagi SLAF |
| < 500 Mb | 100 tys. lub WGS |
| 500 Mb-1 Gb | 100 tys |
| 1 Gb -2 Gb | 200 tys |
| Gigantyczne lub złożone genomy | 300 - 400 tys |
| Aplikacje
| Zalecana Skala populacji
| Strategia sekwencjonowania i głębokość
| |
| Głębokość
| Numer znacznika
| ||
| GWAS
| Liczba próbek ≥ 200
| 10X
|
Według wielkość genomu
|
| Ewolucja genetyczna
| Osoby z każdego podgrupa ≥ 10; próbki ogółem ≥ 30
| 10X
| |
Zalecana dostawa próbek
Pojemnik: probówka wirówkowa o pojemności 2 ml
W przypadku większości próbek nie zalecamy konserwowania w etanolu.
Etykietowanie próbek: Próbki muszą być wyraźnie oznakowane i identyczne z przesłanymi formularzami informacyjnymi.
Wysyłka: Suchy lód: Próbki należy najpierw zapakować w worki i zakopać w suchym lodzie.
Przepływ pracy w serwisie






Próbka kontroli jakości
Eksperyment pilotażowy
Eksperyment SLAF
Przygotowanie biblioteki
Sekwencjonowanie
Analiza danych
Usługi posprzedażowe
1. Statystyka wyniku mapy

2. Rozwój markera SLAF

3. Adnotacja o odmianie

| Rok | Dziennik | IF | Tytuł | Aplikacje |
| 2022 | Komunikacja przyrodnicza | 17.694 | Genomowe podstawy giga-chromosomów i giga-genomu piwonii drzewiastej Paeonia ostii | SLAF-GWAS |
| 2015 | Nowy fitolog | 7.433 | Ślady udomowienia zakotwiczają regiony genomowe o znaczeniu rolniczym w soja | SLAF-GWAS |
| 2022 | Journal of Advanced Research | 12.822 | Sztuczne introgresje Gossypium barbadense obejmujące cały genom do G. hirsutum ujawniają doskonałe loci dla jednoczesnej poprawy jakości i wydajności włókien bawełnianych cechy | SLAF – genetyka ewolucyjna |
| 2019 | Roślina molekularna | 10.81 | Analiza genomu populacji i zgromadzenie De Novo ujawniają pochodzenie chwastów Ryż jako gra ewolucyjna | SLAF – genetyka ewolucyjna |
| 2019 | Genetyka natury | 31.616 | Sekwencja genomu i różnorodność genetyczna karpia pospolitego Cyprinus carpio | Mapa powiązań SLAF |
| 2014 | Genetyka natury | 25.455 | Genom uprawianych orzeszków ziemnych zapewnia wgląd w kariotypy roślin strączkowych, poliploidalne ewolucja i udomowienie roślin. | Mapa powiązań SLAF |
| 2022 | Dziennik biotechnologii roślin | 9.803 | Identyfikacja ST1 ujawnia selekcję obejmującą autostop w zakresie morfologii nasion i zawartości oleju podczas udomowienia soi | Rozwój znacznika SLAF |
| 2022 | International Journal of Molecular Sciences | 6.208 | Identyfikacja i rozwój markerów DNA dla pszenicy-Leymus mollis 2Ns (2D) Disomiczna substytucja chromosomu | Rozwój znacznika SLAF |





