

Sekwencjonowanie genomu roślin/zwierząt de novo




Zalety serwisu
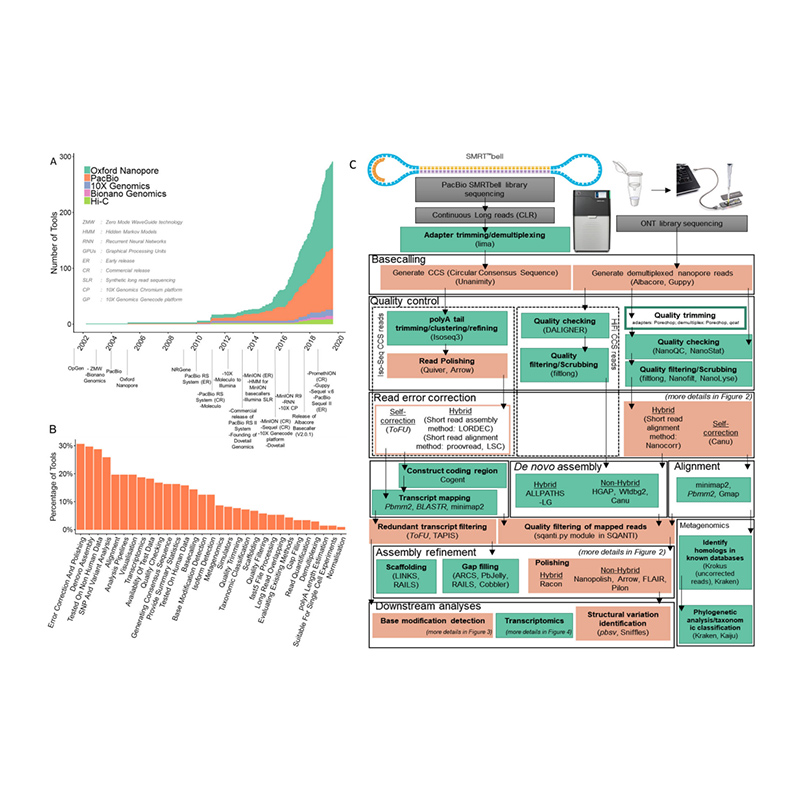
Rozwój platform sekwencjonowania i bioinformatyki wod nowaskładanie genomu
(Amarasinghe SL i in.,Biologia genomu, 2020)
● Konstruowanie nowych genomów i ulepszanie istniejących genomów referencyjnych dla interesujących gatunków.
● Większa dokładność, ciągłość i kompletność montażu
● Tworzenie podstawowych zasobów do badań nad polimorfizmem sekwencji, QTL, edycją genów, hodowlą itp.
● Wyposażone w pełne spektrum platform sekwencjonowania trzeciej generacji: kompleksowe rozwiązanie do składania genomu
● Elastyczne strategie sekwencjonowania i składania spełniające różnorodne genomy o różnych cechach
● Wysoko wykwalifikowany zespół bioinformatyków z dużym doświadczeniem w złożonych składach genomu, w tym poliploidalnych, gigantycznych genomach itp.
● Ponad 100 zakończonych sukcesem spraw, których skumulowany opublikowany współczynnik wpływu wynosi ponad 900
● Czas realizacji wynoszący zaledwie 3 miesiące w przypadku składania genomu na poziomie chromosomu.
● Solidne wsparcie techniczne z szeregiem patentów i praw autorskich do oprogramowania, zarówno po stronie eksperymentalnej, jak i bioinformatycznej.
Specyfikacje usług
|
Treść
|
Platforma
|
Przeczytaj Długość
|
Zasięg
|
| Badanie genomu
| Illumina NovaSekw
| PE150
| ≥ 50X
|
| Sekwencjonowanie genomu
| PacBio Revio
| 15 KB odczytów HiFi
| ≥ 30X
|
| Hi-C
| Illumina NovaSekw
| PE150
| ≥100X
|
Przepływ pracy

Przykładowe wymagania i dostawa
Przykładowe wymagania:
| Gatunek | Tkanka | Dla PacBio | Dla Nanoporu |
| Zwierząt | Narządy trzewne (wątroba, śledziona itp.) | ≥ 1,0 g | ≥ 3,5 g |
| Mięsień | ≥ 1,5 g | ≥ 5,0 g | |
| Krew ssaków | ≥ 1,5 ml | ≥ 5,0 ml | |
| Krew ryb lub ptaków | ≥ 0,2 ml | ≥ 0,5 ml | |
| Rośliny | Świeże liście | ≥ 1,5 g | ≥ 5,0 g |
| Płatek lub łodyga | ≥ 3,5 g | ≥ 10,0 g | |
| Korzenie lub nasiona | ≥ 7,0 g | ≥ 20,0 g | |
| Komórki | Hodowlę komórkową | ≥ 3×107 | ≥ 1×108 |
Zalecana dostawa próbek
Pojemnik: probówka wirówkowa o pojemności 2 ml (nie zaleca się stosowania folii aluminiowej)
W przypadku większości próbek nie zalecamy konserwowania w etanolu.
Etykietowanie próbek: Próbki muszą być wyraźnie oznakowane i identyczne z przesłanymi formularzami informacyjnymi.
Wysyłka: Suchy lód: Próbki należy najpierw zapakować w worki i zakopać w suchym lodzie.
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja DNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
*Pokazane tutaj wyniki demonstracyjne pochodzą z genomów opublikowanych przez Biomarker Technologies
1.Circos na temat składania genomu na poziomie chromosomuG. rotundifoliumprzez platformę sekwencjonowania Nanopore
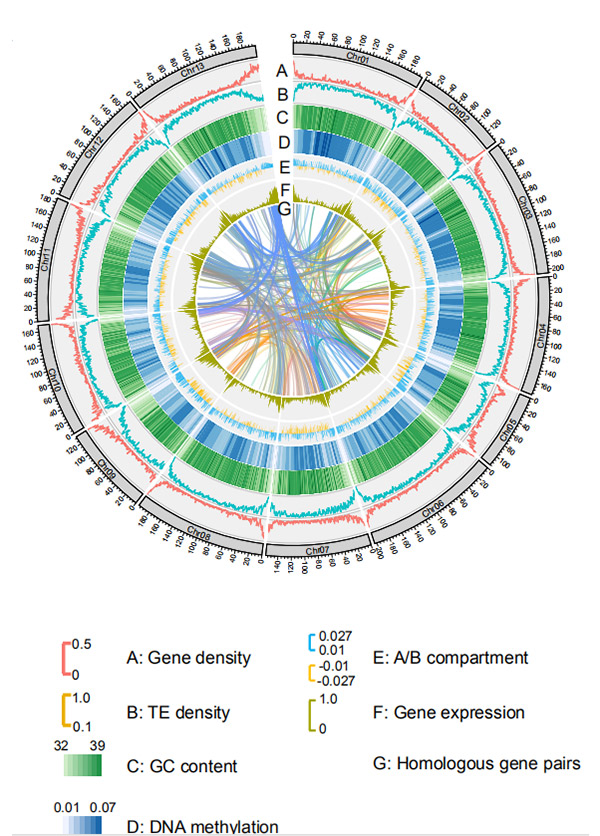
Wang M. i in.,Biologia molekularna i ewolucja, 2021
2.Statystyka składania genomu żyta Weining i adnotacja
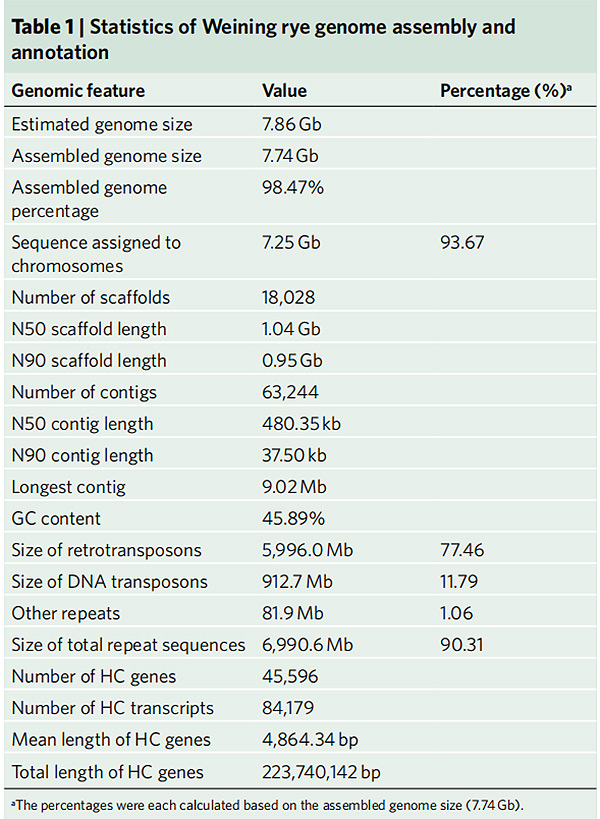
Li G i in.,Genetyka natury, 2021
3. Przewidywanie genówEdule Sechiumgenom, uzyskany na podstawie trzech metod przewidywania:Od nowaprzewidywanie, przewidywanie oparte na homologii i przewidywanie oparte na danych RNA-Seq
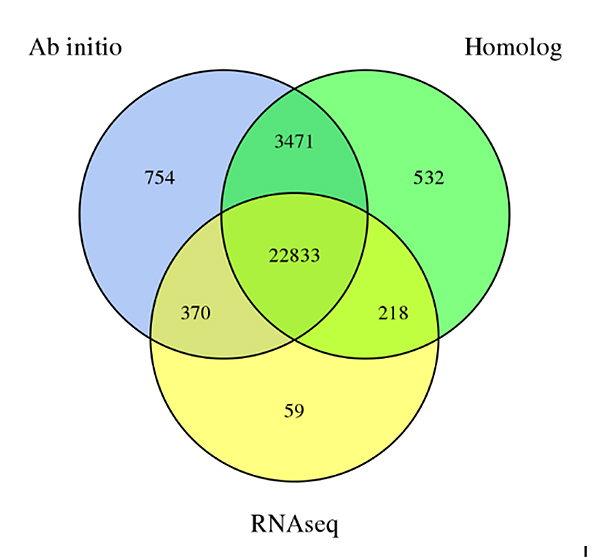
Fu A i in.,Badania ogrodnicze, 2021
4.Identyfikacja nienaruszonych długich powtórzeń końcowych w trzech genomach bawełny
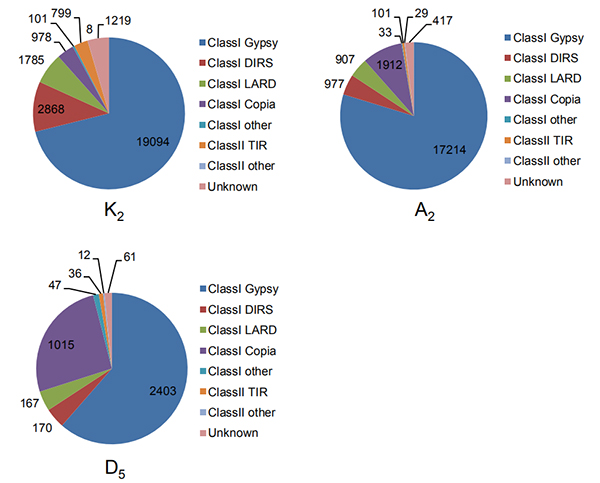
Wang M. i in.,Biologia molekularna i ewolucja, 2021
5. Mapa cieplna Hi-CC. acuminatagenom pokazujący kompleksowe interakcje obejmujące cały genom.Intensywność oddziaływań Hi-C jest proporcjonalna do liniowej odległości pomiędzy kontigami.Czysta linia prosta na tej mapie cieplnej wskazuje na bardzo dokładne zakotwiczenie kontigów na chromosomach.(Współczynnik zakotwiczenia Contig: 96,03%)
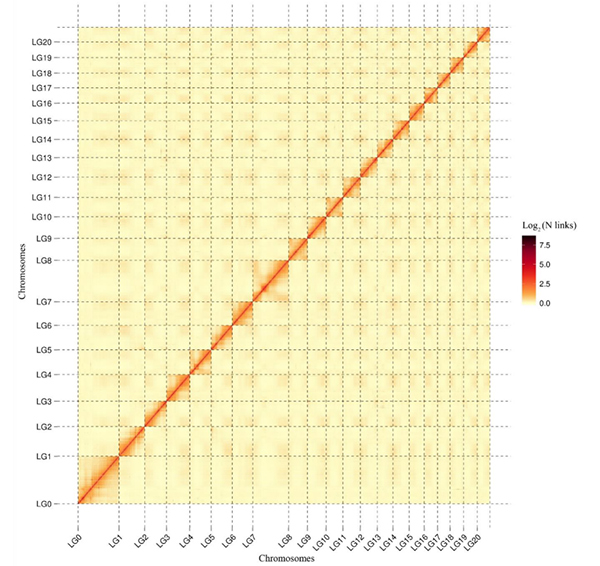
kang M i in.,Komunikacja przyrodnicza,2021
Sprawa BMK
Wysokiej jakości zespół genomu podkreśla cechy genomiczne żyta i geny ważne z agronomicznego punktu widzenia
Opublikowany: Genetyka natury, 2021
Strategia sekwencjonowania:
Składanie genomu: tryb PacBio CLR z biblioteką 20 kb (497 Gb, ok. 63×)
Korekcja sekwencji: NGS z biblioteką DNA o długości 270 bp (430 Gb, ok. 54×) na platformie Illumina
Zakotwiczenie Contigs: biblioteka Hi-C (560 Gb, ok. 71×) na platformie Illumina
Mapa optyczna: (779,55 Gb, ok. 99×) na Bionano Irys
Kluczowe wyniki
1. Opublikowano zestaw genomu żyta Weininga o całkowitej wielkości genomu wynoszącej 7,74 Gb (98,74% szacowanej wielkości genomu metodą cytometrii przepływowej).Rusztowanie N50 tego zespołu osiągnęło 1,04 Gb.93,67% kontigów zostało pomyślnie zakotwiczonych na 7 pseudochromosomach.Zespół ten został oceniony za pomocą mapy powiązań, LAI i BUSCO, co zaowocowało wysokimi wynikami we wszystkich ocenach.
2. Na podstawie tego genomu przeprowadzono dalsze badania z zakresu genomiki porównawczej, mapy powiązań genetycznych, badań transkryptomicznych.Ujawniono szereg cech genomicznych związanych z cechami, w tym duplikacje genów w całym genomie i ich wpływ na geny biosyntezy skrobi;fizyczna organizacja złożonych loci prolaminy, cechy ekspresji genów leżące u podstaw cechy wczesnej głowy oraz domniemane regiony chromosomalne i loci związane z udomowieniem u żyta.
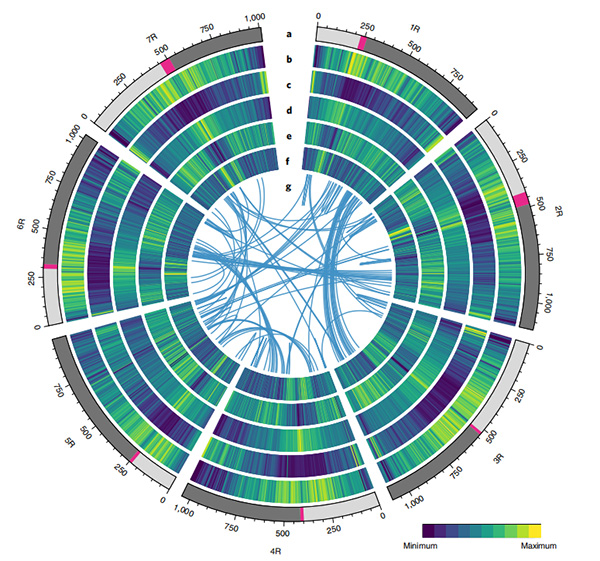 Diagram Circos przedstawiający cechy genomowe genomu żyta Weining | 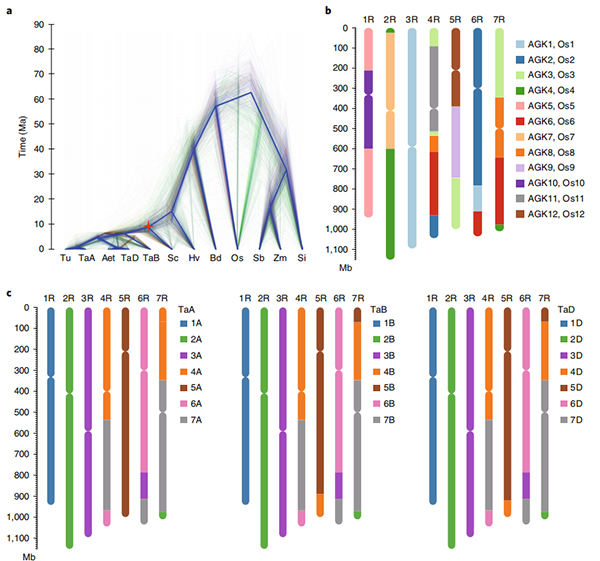 Analizy ewolucyjne i synteny chromosomów genomu żyta |
Li, G., Wang, L., Yang, J.i in.Wysokiej jakości zespół genomu podkreśla cechy genomiczne żyta i geny ważne z agronomicznego punktu widzenia.Nat Genet 53,574–584 (2021).
https://doi.org/10.1038/s41588-021-00808-z





