

Montaż genomu oparty na Hi-C

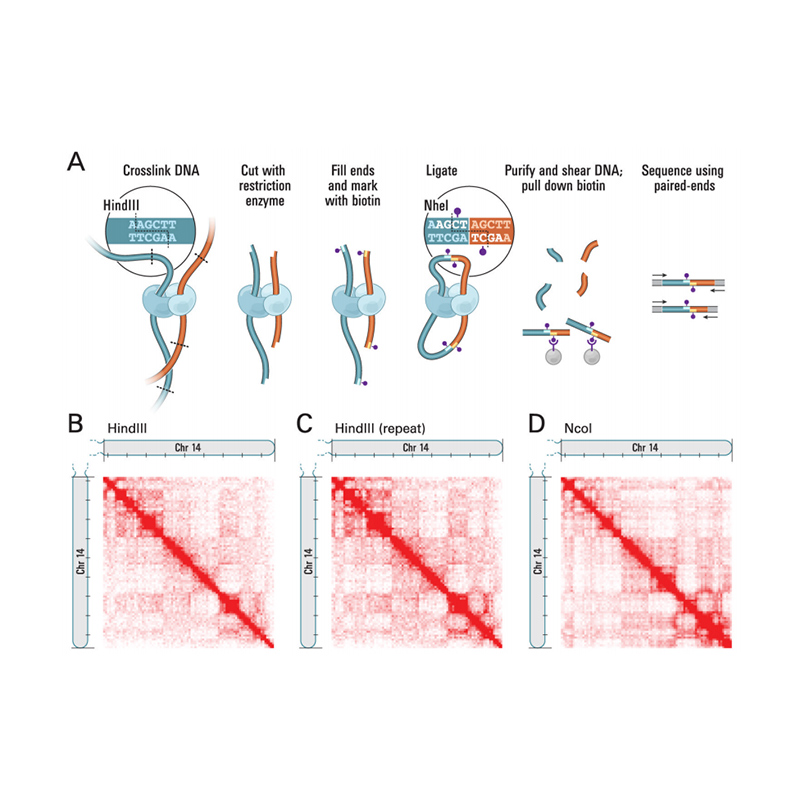






Zalety serwisu
Przegląd Hi-C
(Lieberman-Aiden E i in.,Nauka, 2009)
● Nie ma potrzeby konstruowania populacji genetycznej w celu zakotwiczenia kontigu;
● Większa gęstość znaczników prowadząca do wyższego współczynnika zakotwiczenia kontigów powyżej 90%;
● Umożliwia ocenę i poprawki istniejących zespołów genomu;
● Krótszy czas realizacji i większa dokładność składania genomu;
● Bogate doświadczenie z ponad 1000 bibliotek Hi-C zbudowanych dla ponad 500 gatunków;
● Ponad 100 zakończonych sukcesem przypadków, w których skumulowany opublikowany współczynnik wpływu wynosi ponad 760;
● Składanie genomu w oparciu o Hi-C dla genomu poliploidalnego, w poprzednim projekcie osiągnięto 100% współczynnik zakotwiczenia;
● Własne patenty i prawa autorskie do oprogramowania do eksperymentów Hi-C i analizy danych;
● Opracowane samodzielnie oprogramowanie do wizualizacji danych dostrajających, umożliwia ręczne przesuwanie, cofanie, cofanie i ponawianie bloków.
Specyfikacje usług
|
Typ biblioteki
|
Platforma | Przeczytaj Długość | Poleć strategię |
| Hi-C | Illumina NovaSekw | PE150 | ≥ 100X |
Analizy bioinformatyczne
● Kontrola jakości surowych danych
● Kontrola jakości biblioteki Hi-C
● Składanie genomu w oparciu o Hi-C
● Ocena po montażu
Przykładowe wymagania i dostawa
Przykładowe wymagania:
| Zwierzę | Grzyb | Rośliny
|
| Zamrożona tkanka: 1-2 g na bibliotekę Komórki: 1x 10^7 komórek na bibliotekę | Zamrożona tkanka: 1 g na bibliotekę | Zamrożona tkanka: 1-2 g na bibliotekę
|
| *Zdecydowanie zalecamy wysłanie co najmniej 2 porcji (1 g każda) do eksperymentu Hi-C. | ||
Zalecana dostawa próbek
Pojemnik: probówka wirówkowa o pojemności 2 ml (nie zaleca się stosowania folii aluminiowej)
W przypadku większości próbek nie zalecamy konserwowania w etanolu.
Etykietowanie próbek: Próbki muszą być wyraźnie oznakowane i identyczne z przesłanymi formularzami informacyjnymi.
Wysyłka: Suchy lód: Próbki należy najpierw zapakować w worki i zakopać w suchym lodzie.
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja DNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
*Pokazane tutaj wyniki demonstracyjne pochodzą z genomów opublikowanych przez Biomarker Technologies
1. Mapa cieplna interakcji Hi-CCamptotheca acuminatagenom.Jak pokazano na mapie, intensywność interakcji jest ujemnie skorelowana z odległością liniową, co wskazuje na bardzo dokładne składanie na poziomie chromosomów.(Współczynnik zakotwiczenia: 96,03%)
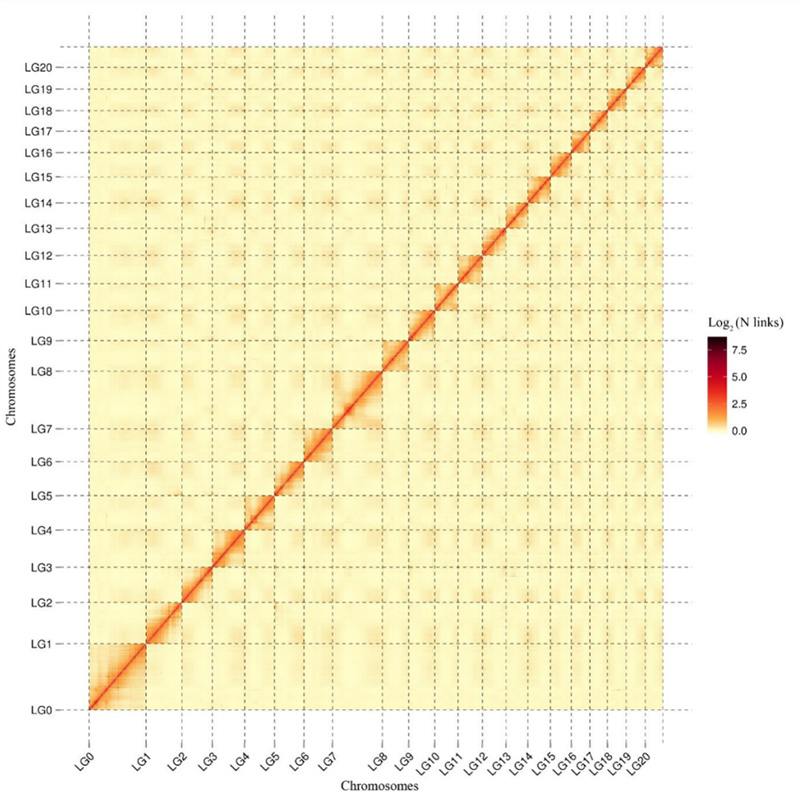
Kang M i in.,Komunikacja przyrodnicza, 2021
2.Hi-C ułatwiło walidację inwersji pomiędzyGossypium hirsutumL. TM-1 A06 iG. arboreumChr06
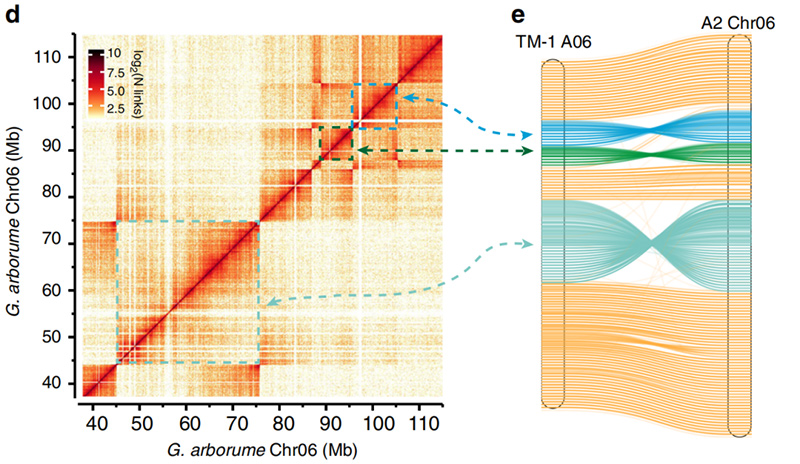
Yang Z i in.,Komunikacja przyrodnicza, 2019
3.Składanie i bialleliczne różnicowanie genomu manioku SC205.Mapa termiczna Hi-C pokazuje wyraźny podział na homologiczne chromosomy.
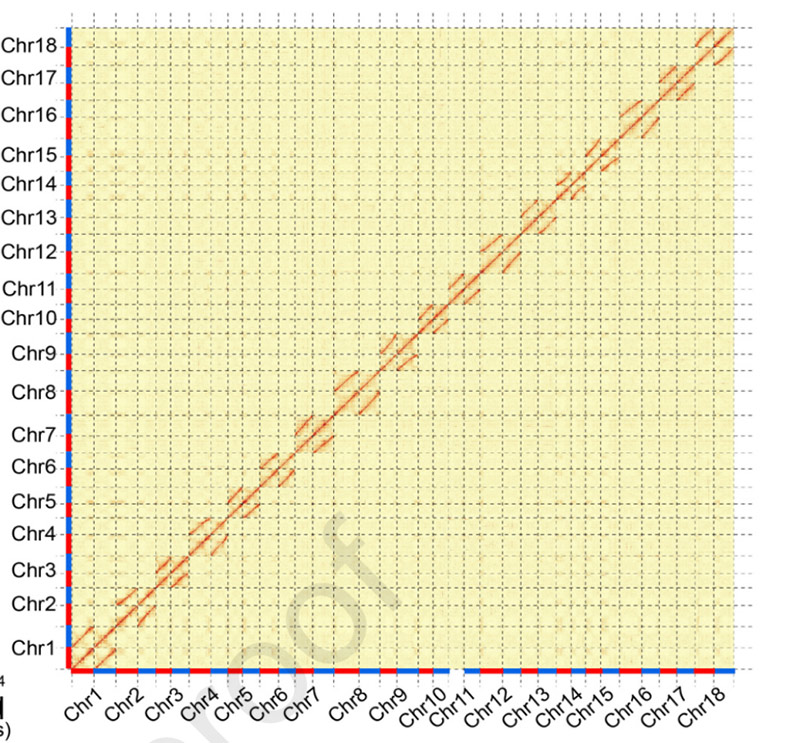
Hu W i in.,Roślina molekularna, 2021
4. Mapa cieplna Hi-C na zespole genomu dwóch gatunków Ficus:F.microcarpa(współczynnik zakotwiczenia: 99,3%) iF.hispida (współczynnik zakotwiczenia: 99,7%)
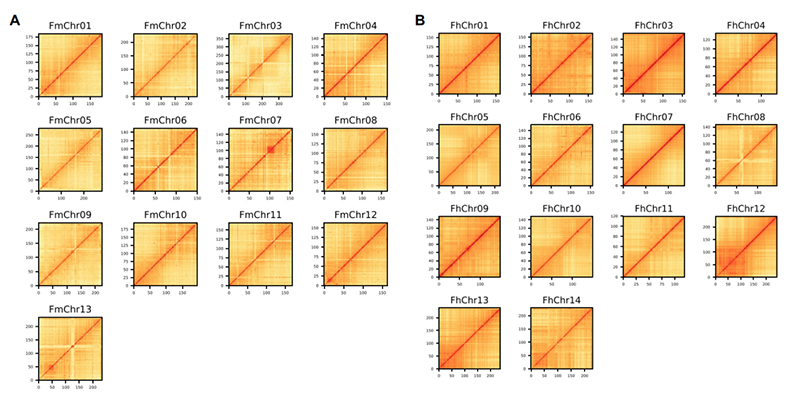
Zhang X i in.,Komórka, 2020
Sprawa BMK
Genomy figowca i osy zapylającej dostarczają wglądu w koewolucję figowo-osy
Opublikowany: Komórka, 2020
Strategia sekwencjonowania:
F. microcarpa genom: ok.84 X PacBio RSII (36,87 Gb) + Hi-C (44 Gb)
F. hispidagenom: ok.97 X PacBio RSII (36,12 Gb) + Hi-C (60 Gb)
Eupristina verticillatagenom: ok.170 X PacBio RSII (65 Gb)
Kluczowe wyniki
1. Skonstruowano dwa genomy drzewa figowego i jeden genom osy zapylającej przy użyciu sekwencjonowania PacBio, Hi-C i mapy powiązań.
(1)F. microcarpagenom: Ustalono zbiór o wielkości 426 Mb (97,7% szacowanej wielkości genomu) z kontigiem N50 o wielkości 908 Kb, wynik BUSCO wynoszący 95,6%.W sumie sekwencje o wielkości 423 Mb zakotwiczono do 13 chromosomów za pomocą Hi-C.Adnotacja genomu dała 29 416 genów kodujących białka.
(2)F. Hispidagenom: Uzyskano zbiór 360 Mb (97,3% szacowanej wielkości genomu) z kontigiem N50 wynoszącym 492 Kb i wynikiem BUSCO wynoszącym 97,4%.Łącznie sekwencje o wielkości 359 Mb zakotwiczono na 14 chromosomach za pomocą Hi-C i były one wysoce identyczne z mapą powiązań o dużej gęstości.
(3)Eupristina verticillatagenom: Ustalono zbiór 387 Mb (szacowany rozmiar genomu: 382 Mb) z kontigiem N50 wynoszącym 3,1 Mb i wynikiem BUSCO wynoszącym 97,7%.
2. Porównawcza analiza genomiki ujawniła dużą liczbę różnic w strukturze między nimiFigowiecgenomy, które dostarczyły bezcennego zasobu genetycznego do badań ewolucji adaptacyjnej.Badanie to po raz pierwszy dostarczyło wglądu w koewolucję osy figowej na poziomie genomu.
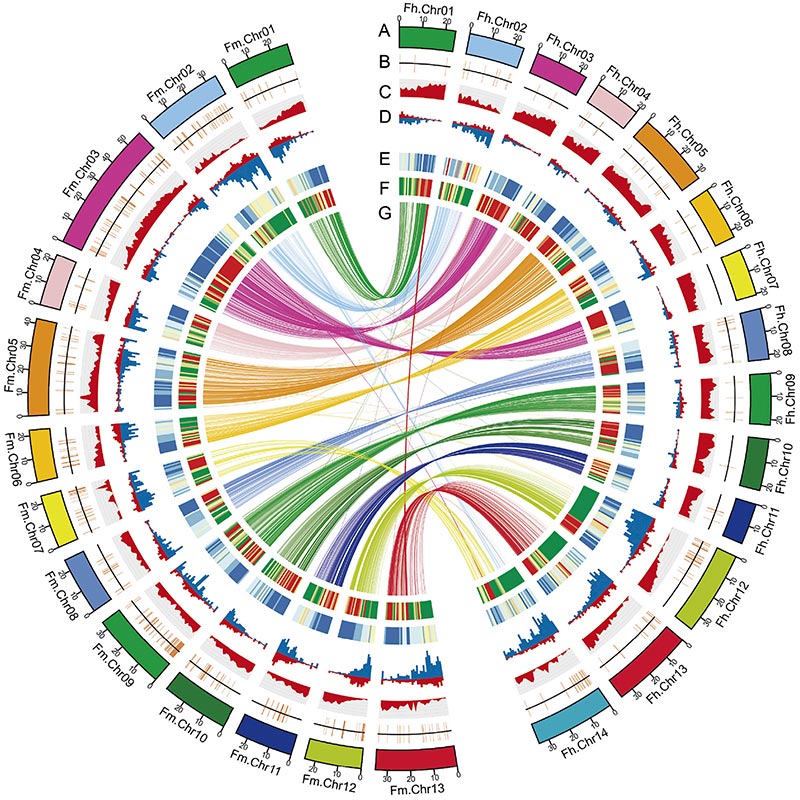 Diagram Circos przedstawiający cechy genomowe dwóchFigowiecgenomy, w tym chromosomy, duplikacje segmentowe (SD), transpozony (LTR, TE, DNA TE), ekspresja i syntenia genów | 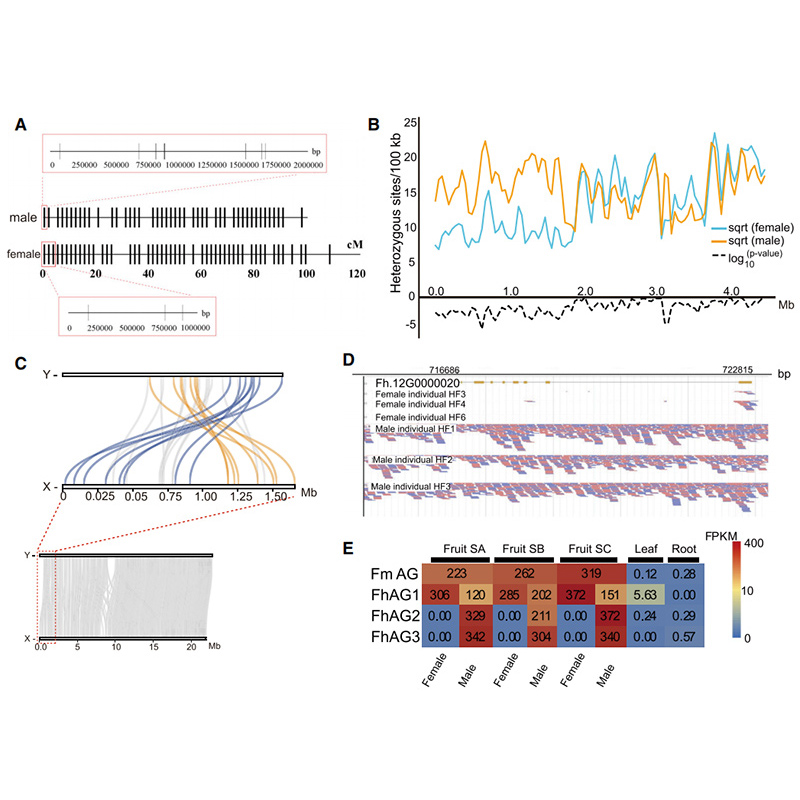 Identyfikacja genu kandydującego na chromosom Y i determinację płci |
Zhang, X. i in.„Genomy drzewa bananowego i osy zapylającej dostarczają wglądu w koewolucję fig i osy”.Komórka 183,4 (2020).





