

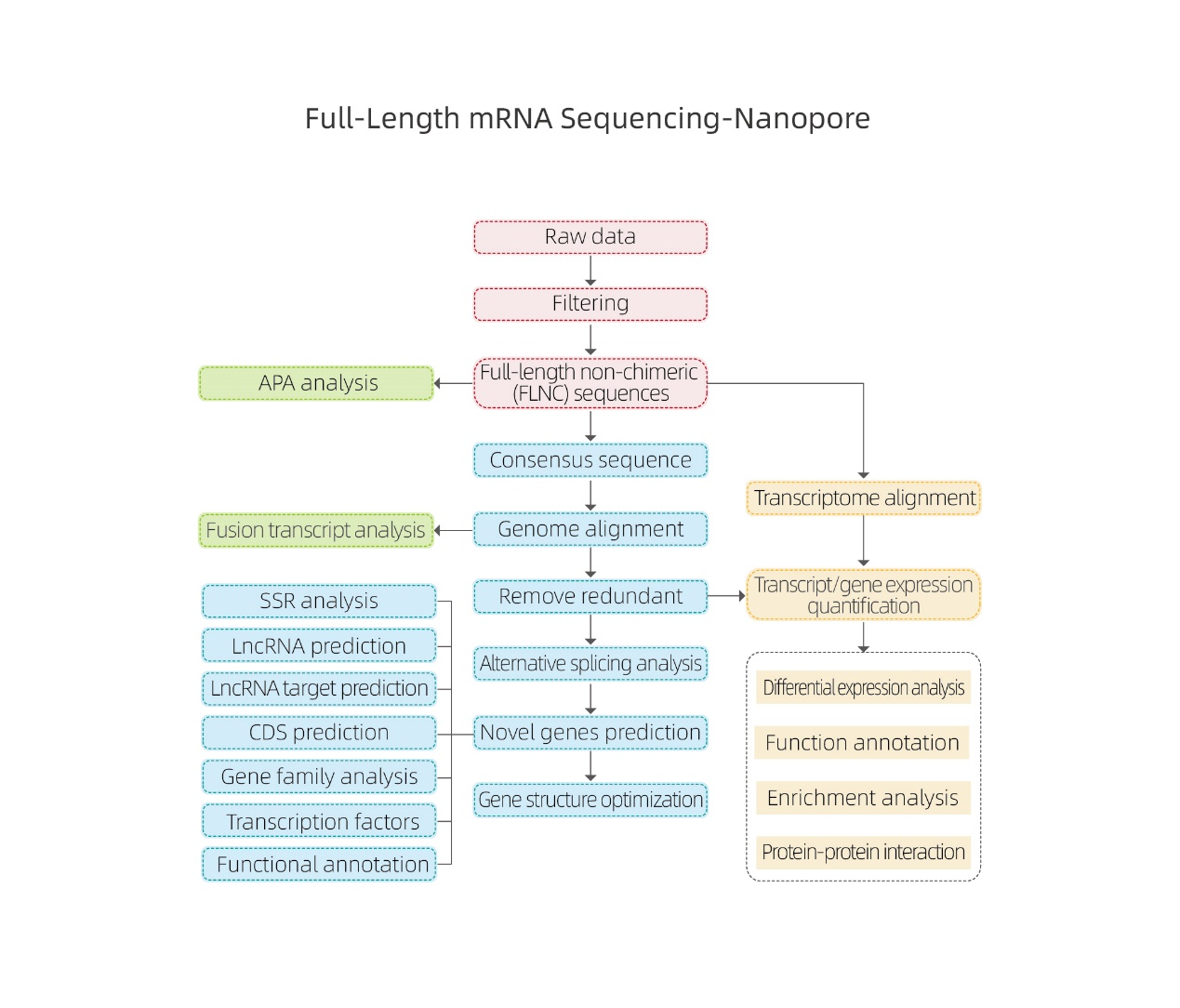
Pełnej długości sekwencjonowanie mRNA-Nanopore





Zalety serwisu
● Odchylenie niskiej sekwencji
● Ujawnianie cząsteczek cDNA pełnej długości
● Mniej danych wymaganych do obsługi tej samej liczby transkrypcji
● Identyfikacja wielu izoform na gen
● Kwantyfikacja ekspresji na poziomie izoform
Specyfikacje usług
| Biblioteka | Platforma | Zalecana wydajność danych (Gb) | Kontrola jakości |
| cDNA-PCR (wzbogacony w Poli-A) | Nanopore PromethION P48 | 6 Gb/próbkę (w zależności od gatunku) | Stosunek pełnej długości> 70% Średni wynik jakości: Q10
|
Analizy bioinformatyczne
●Przetwarzanie surowych danych
● Identyfikacja transkrypcji
● Alternatywne łączenie
● Kwantyfikacja ekspresji na poziomie genu i poziomie izoformy
● Analiza wyrażeń różniczkowych
● Adnotacje i wzbogacanie funkcji (DEG i DET)
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Nukleotydy:
| Stężenie (ng/μl) | Ilość (µg) | Czystość | Uczciwość |
| ≥ 100 | ≥ 0,6 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Na żelu widoczne jest ograniczone lub żadne zanieczyszczenie białkiem lub DNA. | Dla roślin: RIN≥7,0; Dla zwierząt: RIN≥7,5; 5,0 ≥ 28 S/18 S ≥ 1,0; ograniczone lub żadne wzniesienie linii bazowej |
Tkanka: Waga (sucha): ≥1 g
*W przypadku tkanki o masie mniejszej niż 5 mg zalecamy przesłanie próbki tkanki zamrożonej w ciekłym azocie.
Zawiesina komórek: Liczba komórek = 3 x 1006- 1×107
*Zalecamy wysyłkę zamrożonego lizatu komórkowego.W przypadku, gdy liczba komórek jest mniejsza niż 5 × 105zaleca się błyskawiczne zamrożenie w ciekłym azocie, co jest preferowane w przypadku mikroekstrakcji.
Próbki krwi: Objętość ≥1 ml
Zalecana dostawa próbek
Pojemnik: probówka wirówkowa o pojemności 2 ml (nie zaleca się stosowania folii aluminiowej)
Przykładowe oznakowanie: Grupa+replika, np. A1, A2, A3;B1, B2, B3... ...
Wysyłka: 2、Suchy lód: Próbki należy zapakować w worki i zakopać w suchym lodzie.
- Probówki RNAstable: Próbki RNA można suszyć w probówkach do stabilizacji RNA (np. RNAstable®) i przesyłać w temperaturze pokojowej.
Przebieg prac serwisowych
Nukleotydy:

Dostawa próbek

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
Przebieg prac serwisowych
Tkanka:

Projekt eksperymentu
Dostawa próbek

Ekstrakcja RNA
Budowa biblioteki
Sekwencjonowanie
Analiza danych
Usługi posprzedażowe
1.Analiza wyrażeń różniczkowych -Wykres wulkanu
Analizę ekspresji różnicowej można przeprowadzić zarówno na poziomie genów, aby zidentyfikować geny ulegające różnej ekspresji (DEG), jak i na poziomie izoform, aby zidentyfikować je w różny sposób
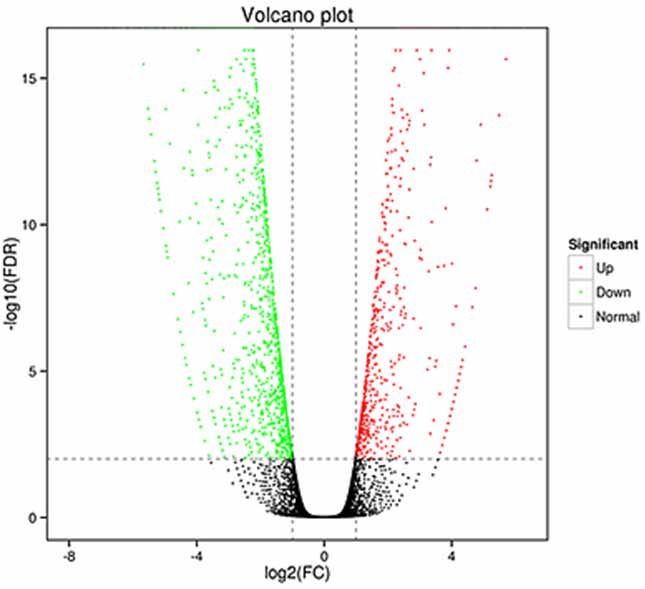
wyrażone transkrypty (DET)
2.Hierarchiczna mapa cieplna grupowania

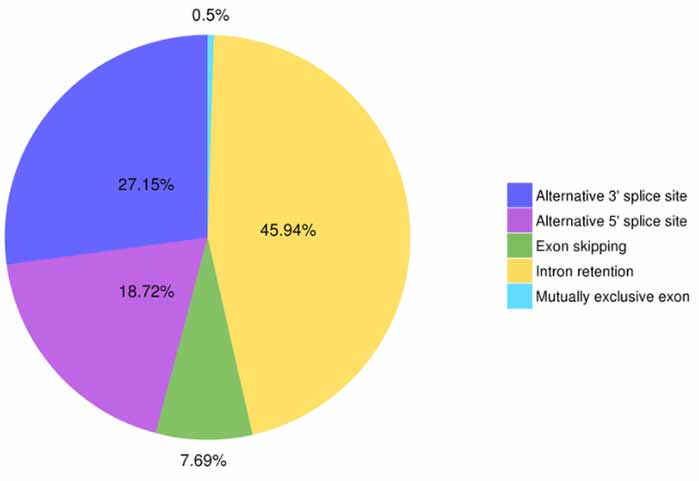
3. Identyfikacja i klasyfikacja alternatywnego splicingu
Astalavista może przewidzieć pięć typów zdarzeń alternatywnego splicingu.
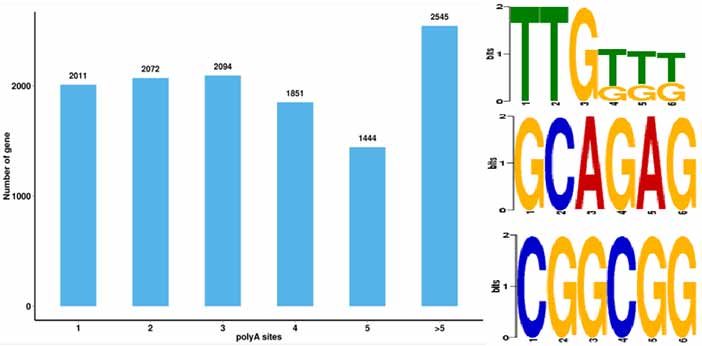
4.Identyfikacja alternatywnych zdarzeń poliadenylacji (APA) i motywu w odległości 50 bp powyżej poli-A
Sprawa BMK
Identyfikacja alternatywnego splicingu i kwantyfikacja na poziomie izoform za pomocą sekwencjonowania transkryptomu pełnej długości nanoporów
Opublikowany:Komunikacja przyrodnicza, 2020
Strategia sekwencjonowania:
Grupowanie: 1. CLL-SF3B1(WT);2. CLL-SF3B1 (mutacja K700E);3. Normalne komórki B
Strategia sekwencjonowania: sekwencjonowanie biblioteki MinION 2D, sekwencjonowanie biblioteki PromethION 1D;krótkiego odczytu danych z tych samych próbek
Platforma sekwencjonowania: Nanopore MinION;Nanoporowy PromethION;
Kluczowe wyniki
1. Identyfikacja alternatywnego splicingu na poziomie izoformy
Sekwencje z długim odczytem umożliwiają identyfikację zmutowanego SF3B1K700E-zmienione miejsca splicingu na poziomie izoformy.Stwierdzono, że 35 alternatywnych 3′SS i 10 alternatywnych 5′SS ma znacząco zróżnicowany splicing pomiędzy SF3B1K700Ei SF3B1WT.33 z 35 zmian zostało nowo odkrytych na podstawie długo czytanych sekwencji.
2. Kwantyfikacja alternatywnego splicingu na poziomie izoformy
Ekspresja izoform retencji intronów (IR) w SF3B1K700Ei SF3B1WTzostały określone ilościowo na podstawie sekwencji nanoporów, ujawniając globalną regulację w dół izoform IR w SF3B1K700E.
Odniesienie
Tang AD, Soulette CM, Baren MJV i in.Charakterystyka transkryptu pełnej długości mutacji SF3B1 w przewlekłej białaczce limfatycznej ujawnia regulację w dół zatrzymanych intronów [J].Komunikacja przyrodnicza.





