

Sekwencjonowanie mRNA pełnej długości -PacBio





Zalety serwisu
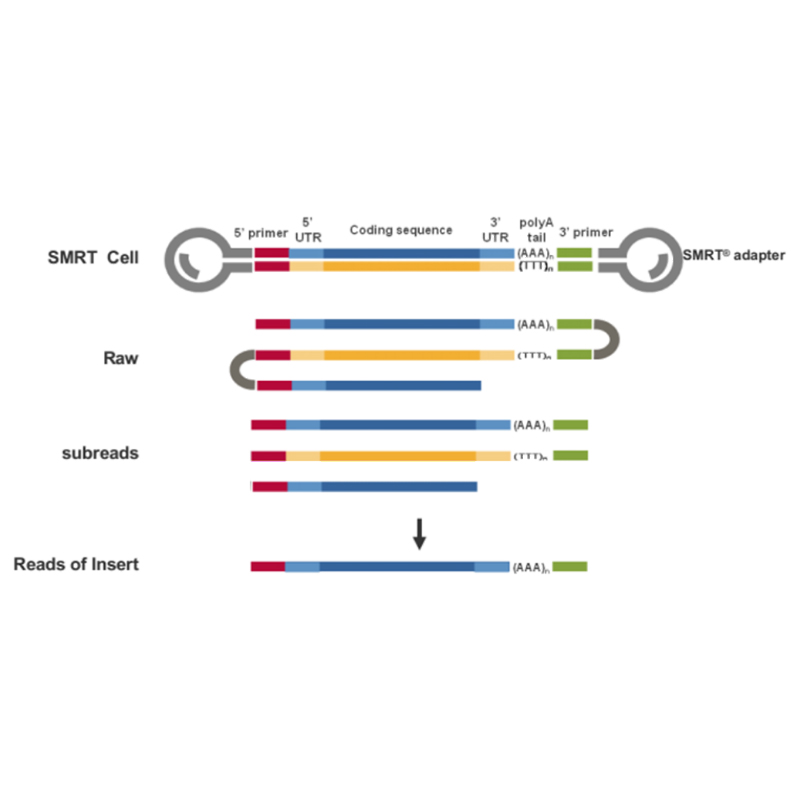
● Bezpośredni odczyt cząsteczki cDNA pełnej długości od końca 3' do końca 5'
● Rozdzielczość na poziomie izoformy w strukturze sekwencji
● Transkrypcje charakteryzujące się dużą dokładnością i integralnością
● Wysoce kompatybilny z gatunkami vaiours
● Duża wydajność sekwencjonowania dzięki wyposażeniu w 4 platformy sekwencjonowania PacBio Sequel II
● Duże doświadczenie w ponad 700 projektach sekwencjonowania RNA w oparciu o Pacbio
● Dostarczanie wyników w oparciu o BMKCloud: Indywidualna eksploracja danych dostępna na platformie.
● Usługi posprzedażowe ważne przez 3 miesiące po zakończeniu projektu
Specyfikacje usług
Platforma: PacBio Sequel II
Biblioteka sekwencjonowania: Biblioteka mRNA wzbogacona w Poli A
Zalecana wydajność danych: 20 Gb/próbkę (w zależności od gatunku)
FLNC (%): ≥75%
*FLNC: Niechimeryczne transkrypty pełnej długości
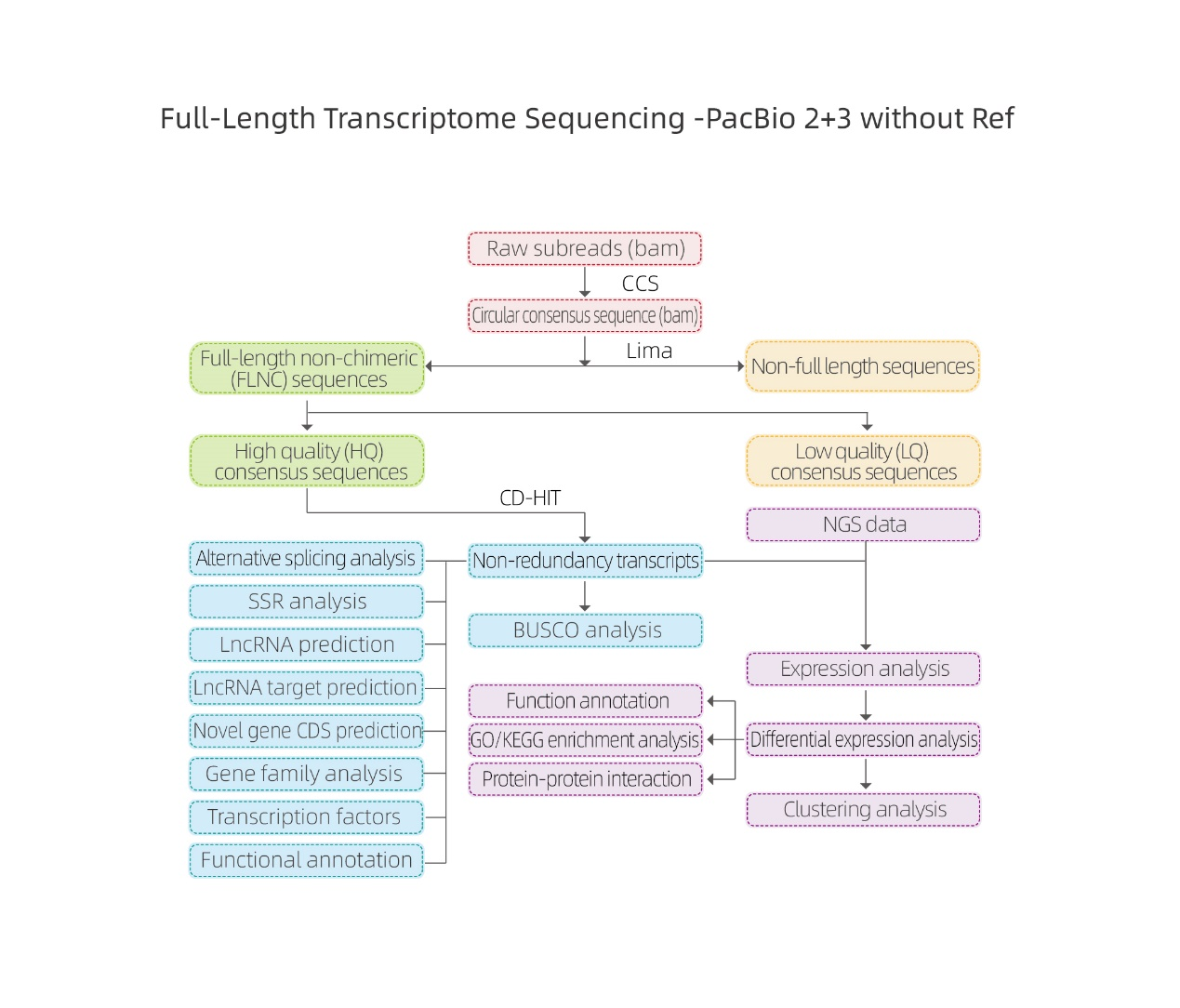
Analizy bioinformatyczne
● Przetwarzanie surowych danych
● Identyfikacja transkrypcji
● Struktura sekwencji
● Kwantyfikacja wyrażeń
● Adnotacja dotycząca funkcji
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Nukleotydy:
| Stężenie (ng/μl) | Ilość (µg) | Czystość | Uczciwość |
| ≥ 120 | ≥ 0,6 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Na żelu widoczne jest ograniczone lub żadne zanieczyszczenie białkiem lub DNA. | Dla roślin: RIN≥7,5; Dla zwierząt: RIN≥8,0; 5,0 ≥ 28 S/18 S ≥ 1,0; ograniczone lub żadne wzniesienie linii bazowej |
Tkanka: Waga (sucha):≥1 g
*W przypadku tkanki o masie mniejszej niż 5 mg zalecamy przesłanie próbki tkanki zamrożonej w ciekłym azocie.
Zawiesina komórkowa:Liczba komórek = 3×106- 1×107
*Zalecamy wysyłkę zamrożonego lizatu komórkowego.W przypadku, gdy liczba komórek jest mniejsza niż 5 × 105zaleca się błyskawiczne zamrożenie w ciekłym azocie, co jest preferowane w przypadku mikroekstrakcji.
Próbki krwi:Objętość ≥1 mL
Mikroorganizm:Masa ≥ 1 g
Zalecana dostawa próbek
Pojemnik:
Probówka wirówkowa o pojemności 2 ml (nie zaleca się stosowania folii aluminiowej)
Przykładowe oznakowanie: Grupa+replika, np. A1, A2, A3;B1, B2, B3... ...
Wysyłka:
1. Suchy lód: Próbki należy zapakować do worków i zakopać w suchym lodzie.
2. Probówki RNAstable: Próbki RNA można suszyć w probówkach do stabilizacji RNA (np. RNAstable®) i przesyłać w temperaturze pokojowej.
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Ekstrakcja RNA

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
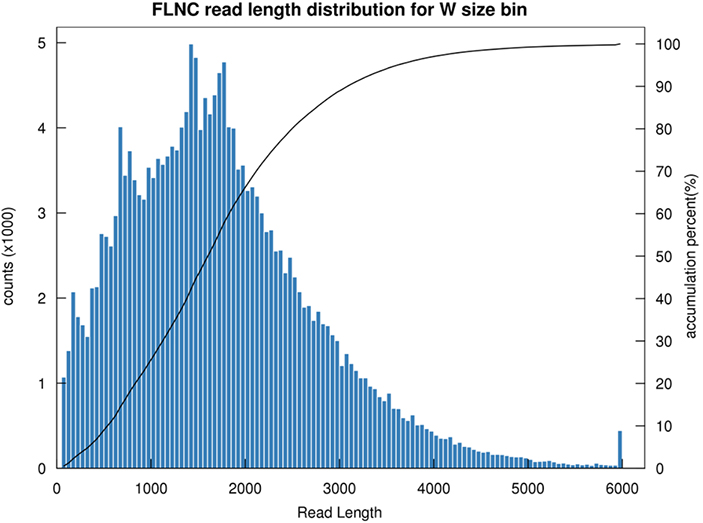
1.Rozkład długości FLNC
Długość niechimerycznego odczytu pełnej długości (FLNC) wskazuje długość cDNA w konstrukcji biblioteki.Rozkład długości FLNC jest kluczowym wskaźnikiem oceny jakości konstrukcji biblioteki.
Rozkład długości odczytu FLNC
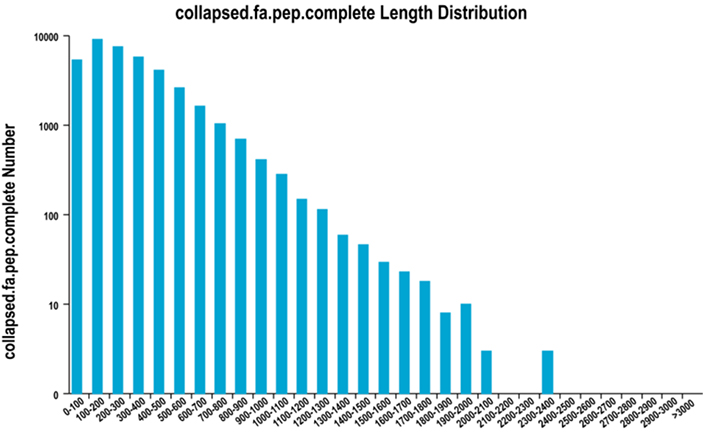
2. Pełny rozkład długości regionu ORF
Używamy TransDecodera do przewidywania regionów kodujących białka i odpowiadających im sekwencji aminokwasów w celu wygenerowania zestawów unigenów, które zawierają pełną, niezbędną informację o transkrypcie we wszystkich próbkach.
Pełny rozkład długości regionu ORF
3.Analiza wzbogacenia szlaku KEGG
Transkrypty o różnej ekspresji (DET) można zidentyfikować, dopasowując dane sekwencjonowania RNA oparte na NGS do zestawów transkryptów pełnej długości wygenerowanych przez dane sekwencjonowania PacBio.Te DET można dalej przetwarzać do różnych analiz funkcjonalnych, np. analizy wzbogacania szlaku KEGG.
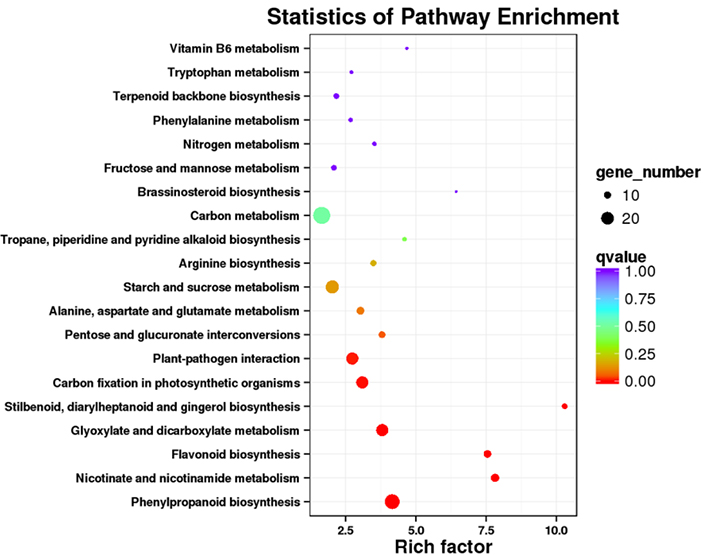
Wzbogacanie szlaku DET KEGG – wykres punktowy
Sprawa BMK
Dynamika rozwoju transkryptomu łodygi Populus
Opublikowany: Dziennik biotechnologii roślin, 2019
Strategia sekwencjonowania:
Przykładowa kolekcja:regiony macierzyste: wierzchołek, pierwszy międzywęzeł (IN1), drugi międzywęzeł (IN2), trzeci międzywęzeł (IN3), międzywęzeł (IN4) i międzywęzeł (IN5) z Nanlin895
Sekwencja NGS:RNA 15 osobników połączono jako jedną próbkę biologiczną.Przetworzono trzy repliki biologiczne każdego punktu pod kątem sekwencji NGS
Sekwencja TGS:Regiony pnia podzielono na trzy regiony, tj. wierzchołek, IN1-IN3 i IN4-IN5.Każdy region poddano sekwencjonowaniu PacBio przy użyciu czterech typów bibliotek: 0-1 kb, 1-2 kb, 2-3 kb i 3-10 kb.
Kluczowe wyniki
1. Łącznie zidentyfikowano 87150 transkryptów pełnej długości, w których zidentyfikowano 2081 nowych izoform i 62058 nowych izoform poddanych alternatywnemu splicingowi.
Zidentyfikowano 2,1187 lncRNA i 356 genów fuzyjnych.
3. Od wzrostu pierwotnego do wzrostu wtórnego zidentyfikowano 15838 transkryptów o różnej ekspresji z 995 genów o różnej ekspresji.We wszystkich DEG 1216 to czynniki transkrypcyjne, z których większość nie została jeszcze opisana.
Analiza wzbogacania 4.GO ujawniła znaczenie podziału komórek i procesu utleniania i redukcji w rozwoju pierwotnym i wtórnym.
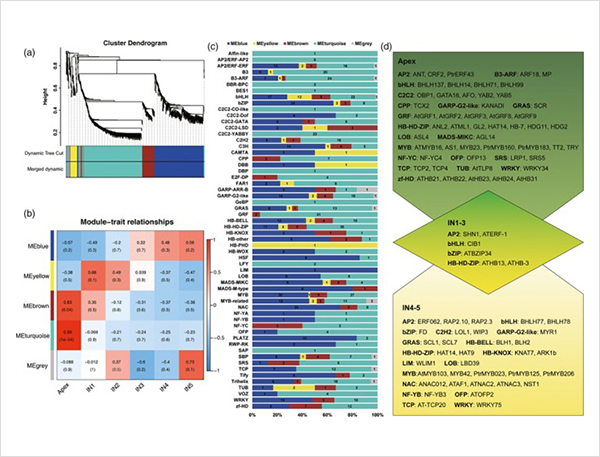
Zdarzenia alternatywnego splicingu i różne izoformy
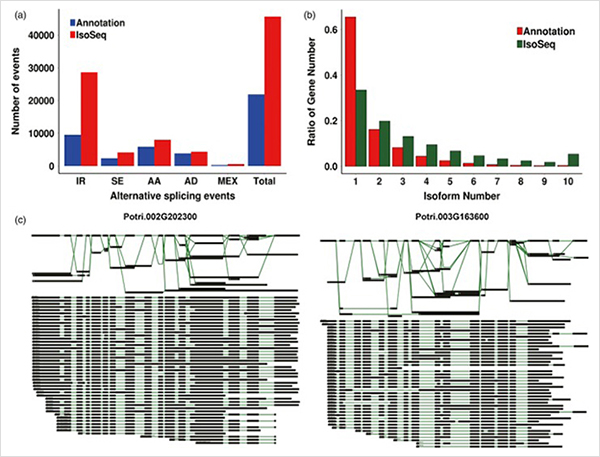
Analiza WGCNA dotycząca czynników transkrypcyjnych
Odniesienie
Chao Q, Gao ZF, Zhang D i in.Dynamika rozwoju transkryptomu łodygi Populus.Plant Biotechnol J. 2019;17(1):206-219.doi:10.1111/pbi.12958





