

Genomika porównawcza






Zalety serwisu
● Kompleksowy pakiet analiz, zawierający osiem najczęściej wymaganych analiz
● Wysoka niezawodność analizy ze szczegółową i łatwo zrozumiałą interpretacją wyników
● Dobrze zaprojektowane, gotowe do publikacji dane liczbowe
● Wysoko wykwalifikowany zespół bioinformatyków spełnia różnorodne, spersonalizowane wymagania w zakresie analiz
● Krótszy czas realizacji i większa dokładność analizy
● Bogate doświadczenie z ponad 90 zakończonymi sukcesem sprawami, w których skumulowany opublikowany współczynnik wpływu wynosi ponad 900
Specyfikacje usług
| Szacowany czas realizacji | Liczba gatunków | Ćwiczenie |
| 30 dni roboczych | 6 - 12 | Grupowanie rodzin genów Rozszerzanie i kurczenie się rodziny genów Konstrukcja drzewa filogenetycznego Oszacowanie czasu rozbieżności (wymagana kalibracja Fossil) Czas wstawienia LTR (dla roślin) Duplikacja całego genomu (dla roślin) Presja selektywna Analiza syntenii |
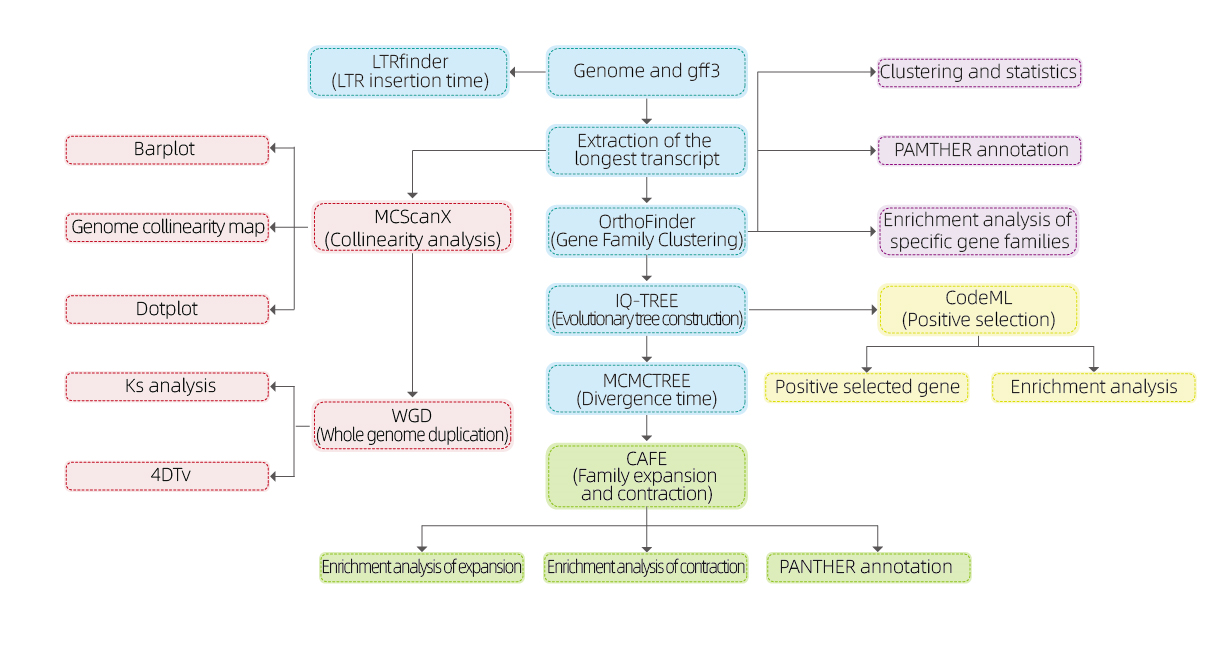
Analizy bioinformatyczne
● Rodzina genów
● Filogenetyka
● Czas rozbieżności
● Ciśnienie selektywne
● Analiza syntenii
Przykładowe wymagania i dostawa
Przykładowe wymagania:
Tkanka lub DNA do sekwencjonowania i składania genomu
Dla tkanki
| Gatunek | Tkanka | Ankieta | PacBio CCS |
| Zwierzę | Tkanka trzewna | 0,5 ~ 1g | ≥ 3,5 g |
| Tkanka mięśniowa | |||
| ≥ 5,0g | |||
| ≥ 5,0 ml | |||
| Krew ssaków | |||
| ≥ 0,5 ml | |||
| Krew drobiowa/rybna | |||
| Zakład | Świeży Liść | 1 ~ 2g | ≥ 5,0g |
| Płatek/łodyga | 1 ~ 2g | ≥ 10,0 g | |
| Korzeń/Nasiono | 1 ~ 2g | ≥ 20,0 g | |
| Komórki | Hodowana komórka | - | ≥ 1 x 108 |
Dane
Pliki sekwencji genomu (.fasta) i pliki adnotacji (.gff3) blisko spokrewnionych gatunków
Przebieg prac serwisowych

Projekt eksperymentu

Dostawa próbek

Budowa biblioteki

Sekwencjonowanie

Analiza danych

Usługi posprzedażowe
*Pokazane tutaj wyniki demonstracyjne pochodzą z genomów opublikowanych przez Biomarker Technologies
1. Oszacowanie czasu insercji LTR: Rysunek pokazuje unikalny rozkład bimodalny czasów insercji LTR-RT w genomie żyta Weining w porównaniu z innymi gatunkami.Ostatni szczyt pojawił się około 0,5 miliona lat temu.
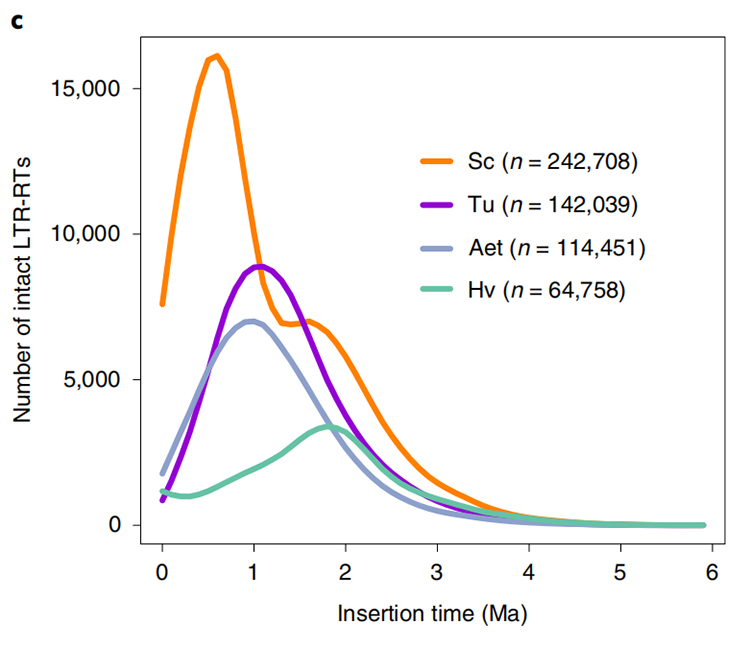
Li Guang i in.,Genetyka natury, 2021
2.Filogeneza i analiza rodziny genów kolczocha (Sechium edule): Analizując kolczoch i pozostałych 13 spokrewnionych gatunków w rodzinie genów, stwierdzono, że kolczoch jest najbliżej spokrewniony z tykwą wężową (Trichosanthes anguina).Kolczoch pochodzący z tykwy wężowej w około 27–45 milionów lat temu, a duplikację całego genomu (WGD) zaobserwowano u kolczota w czasie 25 ± 4 milionów lat, co jest trzecim zdarzeniem WGD u dyniowatych.
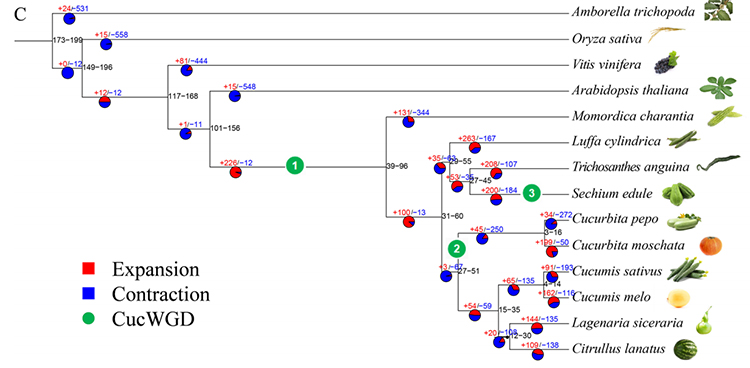
Fu A i in.,Badania ogrodnicze, 2021
3. Analiza synteny: Niektóre geny związane z fitohormonami w rozwoju owoców znaleziono u kolczocha, tykwy wężowej i dyni.Korelacja między kolczochem a dynią jest nieco wyższa niż między kolczochem a tykwą wężową.
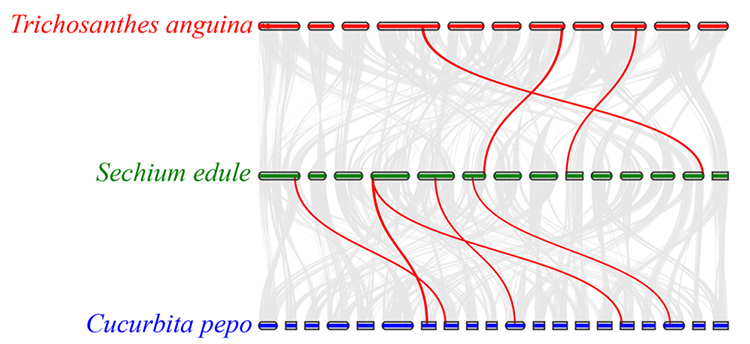
Fu A i in.,Badania ogrodnicze, 2021
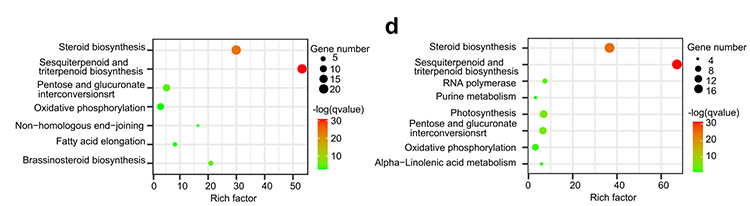
4. Analiza rodziny genów: Wzbogacenie KEGG na ekspansję i kurczenie się rodziny genów w genomach G.thurberi i G.davidsonii wykazało, że geny związane z biosyntezą steroidów i biosyntezą brasinosteroidów uległy ekspansji.
Yang Z i in.,Biologia BMC, 2021
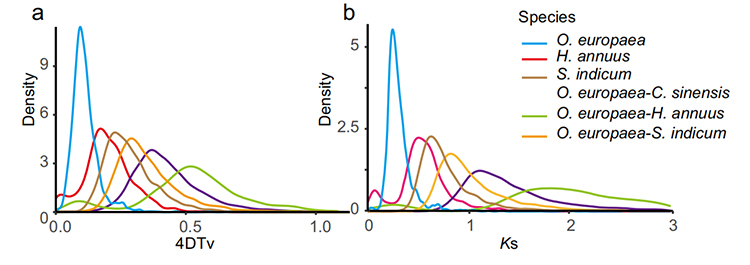
5. Analiza duplikacji całego genomu: Analiza dystrybucji 4DTV i Ks wykazała zdarzenie duplikacji całego genomu.Szczyty wewnątrzgatunkowe wykazały zdarzenia duplikacyjne.Szczyty międzygatunkowe wykazały zdarzenia specjacyjne.Analiza wykazała, że w porównaniu z pozostałymi trzema blisko spokrewnionymi gatunkami, O. europaea niedawno przeszła duplikację genów na dużą skalę.
Rao G i in.,Badania ogrodnicze, 2021
Sprawa BMK
Róża bez kolców: spostrzeżenia genomiczne powiązane z adaptacją do wilgoci
Opublikowany: Narodowy Przegląd Naukowy, 2021
Strategia sekwencjonowania:
„Basye'aBezcierniowy' (R.Wichurain) genom:
Około.93 X PacBio + ok.90 X Nanopor + 267 X Illumina
Kluczowe wyniki
1. Wysokiej jakości genom R.wichuraiana został skonstruowany przy użyciu technik sekwencjonowania o długim odczycie, co dało zbiór o wielkości 530,07 Mb (szacunkowa wielkość genomu wynosiła około 525,9 Mb za pomocą cytometrii przepływowej i 525,5 za pomocą badania genomu; Heterozygotyczność wynosiła około 1,03%).Szacowany wynik BUSCO wyniósł 93,9%.W porównaniu z „Old rumieńcem” (haploOB) jakość i kompletność tego genomu została potwierdzona podstawową dokładnością pojedynczej zasady i wskaźnikiem składania LTR (LAI=20,03).Genom R.wichuraiana zawiera 32 674 genów kodujących białka.
2. Wspólna analiza wieloomiczna, składająca się z genomiki porównawczej, transkryptomiki i analizy QTL populacji genetycznej, ujawniła kluczową specjację pomiędzy R. wichuraiana i Rosa chinensis.Prawdopodobnie zmienność ekspresji pokrewnych genów w QTL była prawdopodobnie powiązana z wzorcem kolców łodygi.
Porównawcza analiza genomiki pomiędzy Basye; Thornless i Rosa chinensis, obejmująca analizę synteny, klaster rodziny genów, analizę ekspansji i kurczenia się, ujawniła dużą liczbę odmian, które były związane z kluczowymi cechami róż.Wyjątkowa ekspansja rodziny genów NAC i FAR1/FRS najprawdopodobniej była powiązana z odpornością na czarną plamistość.
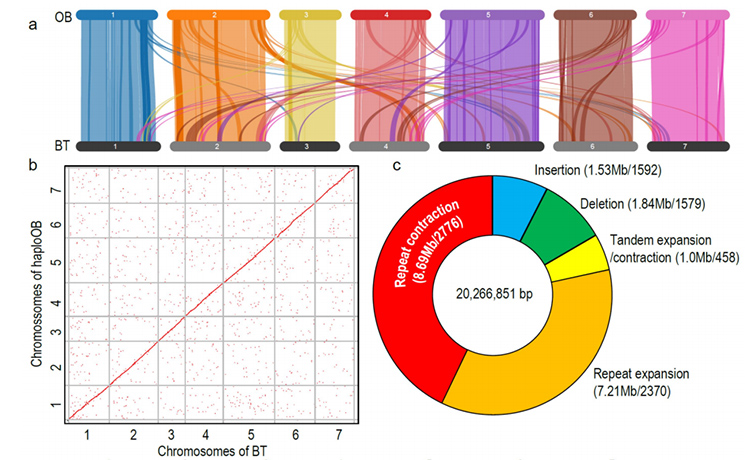

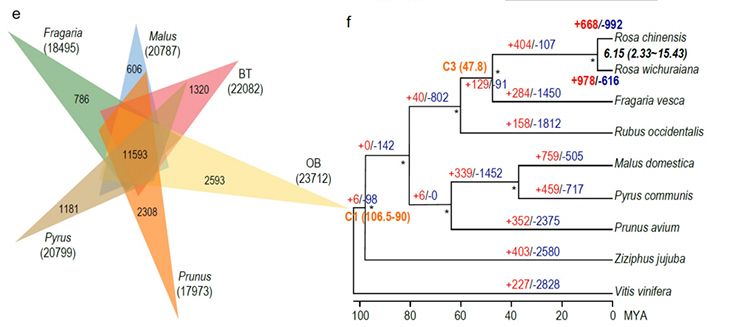
Analiza porównawcza genomiki między genomami BT i haploOB.
Zhong, M. i in.„Róża bez kolców: spostrzeżenia genomiczne powiązane z adaptacją do wilgoci”Narodowy Przegląd Naukowy, 2021;, nwab092.





