

Op Hi-C gebaseerde Genome Assembly







Servicevoordelen
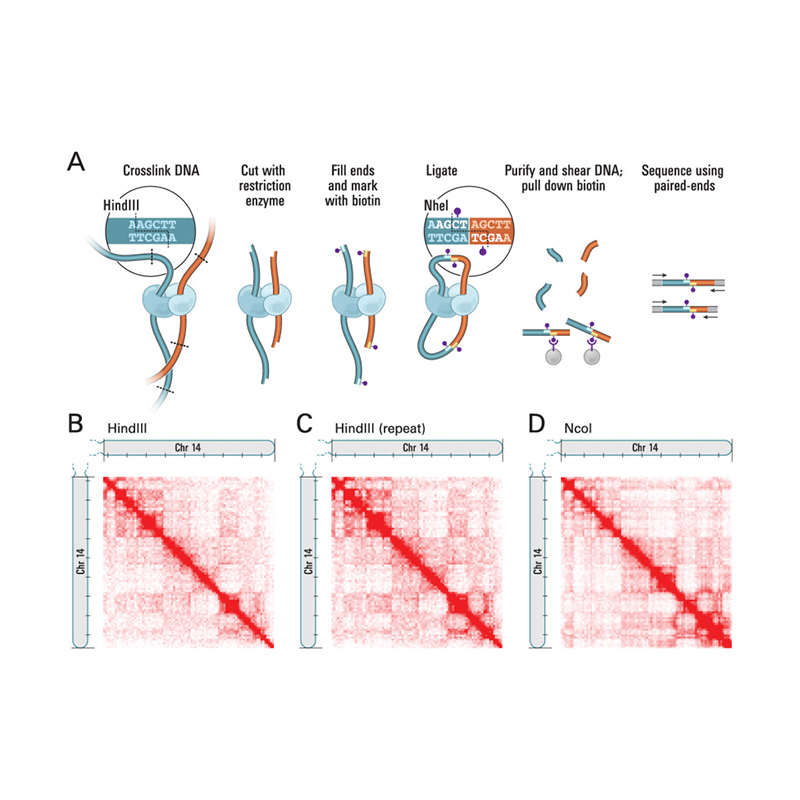
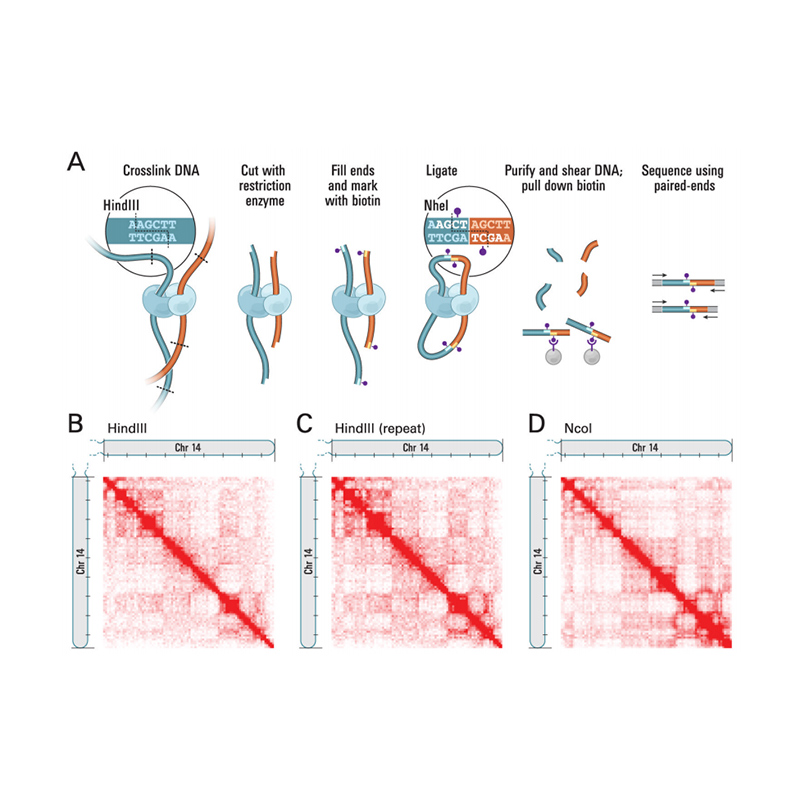
Overzicht van Hi-C
(Lieberman-Aiden E et al.,Wetenschap, 2009)
● Geen noodzaak voor het construeren van een genetische populatie voor contig-verankering;
● Hogere markerdichtheid, wat leidt tot een hogere contigs-verankeringsratio van meer dan 90%;
● Maakt evaluatie en correcties van bestaande genoomassemblages mogelijk;
● Kortere doorlooptijd met hogere nauwkeurigheid bij genoomassemblage;
● Overvloedige ervaring met meer dan 1000 Hi-C-bibliotheken gebouwd voor meer dan 500 soorten;
● Meer dan 100 succesvolle cases met een cumulatieve gepubliceerde impactfactor van meer dan 760;
● Op Hi-C gebaseerde genoomassemblage voor polyploïde genoom, in vorig project werd een verankeringspercentage van 100% bereikt;
● Interne patenten en softwareauteursrechten voor Hi-C-experimenten en data-analyse;
● Zelfontwikkelde software voor het afstemmen van gevisualiseerde gegevens, maakt het handmatig verplaatsen, omkeren, intrekken en opnieuw uitvoeren van blokken mogelijk.
Servicespecificaties
|
Bibliotheektype
|
Platform | Lees lengte | Strategie aanbevelen |
| Hallo-C | Illumina NovaSeq | PE150 | ≥ 100X |
Bio-informatica analyses
● Kwaliteitscontrole van ruwe gegevens
● Kwaliteitscontrole van de Hi-C-bibliotheek
● Op Hi-C gebaseerde genoomassemblage
● Evaluatie na de montage
Monstervereisten en levering
Voorbeeldvereisten:
| Dier | Schimmel | Planten
|
| Bevroren weefsel: 1-2 g per bibliotheek Cellen: 1x 10^7 cellen per bibliotheek | Bevroren weefsel: 1 g per bibliotheek | Bevroren weefsel: 1-2 g per bibliotheek
|
| *We raden ten zeerste aan om ten minste 2 porties (elk 1 g) te sturen voor het Hi-C-experiment. | ||
Aanbevolen monsterlevering
Verpakking: centrifugebuisje van 2 ml (aluminiumfolie wordt niet aanbevolen)
Voor de meeste monsters raden wij aan om ze niet in ethanol te bewaren.
Etikettering van monsters: Monsters moeten duidelijk geëtiketteerd zijn en identiek zijn aan het ingediende monsterinformatieformulier.
Verzending: Droogijs: Monsters moeten eerst in zakken worden verpakt en in droogijs worden begraven.
Servicewerkstroom

Experimentontwerp

Levering van monsters

DNA-extractie

Bouw van bibliotheek

Volgorde aanbrengen in

Gegevensanalyse

After-sales diensten
*De hier getoonde demoresultaten zijn allemaal afkomstig van genomen gepubliceerd met Biomarker Technologies
1.Hi-C interactie warmtekaart vanCamptotheca acuminatagenoom.Zoals op de kaart te zien is, is de intensiteit van interacties negatief gecorreleerd met de lineaire afstand, wat duidt op een zeer nauwkeurige assemblage op chromosoomniveau.(Verankeringsverhouding: 96,03%)
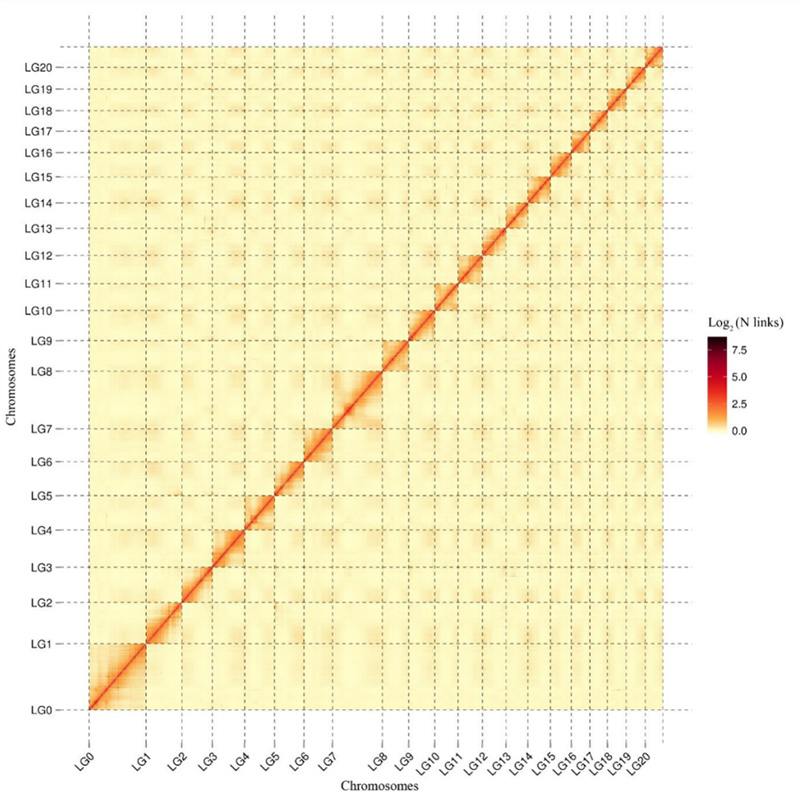
Kang M et al.,Natuurcommunicatie, 2021
2.Hi-C vergemakkelijkte de validatie van inversies tussenGossypium hirsutumL.TM-1 A06 enG. arboreumChr06
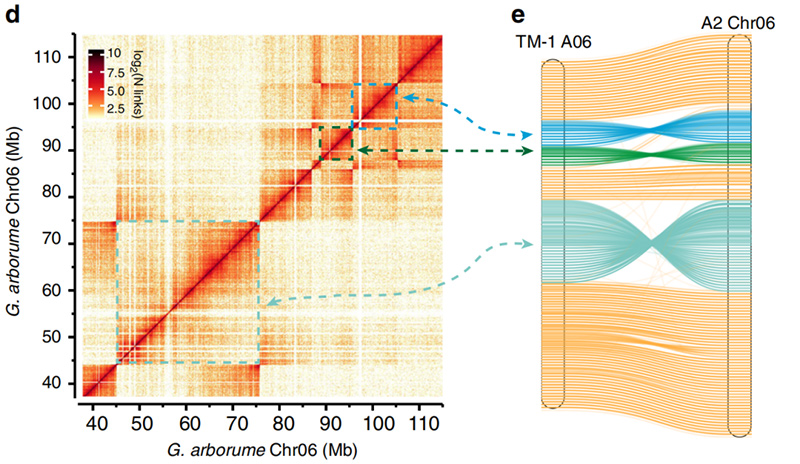
Yang Z et al.,Natuurcommunicatie, 2019
3.Assemblage en biallele differentiatie van het cassavegenoom SC205.Hi-C heatmap vertoonde een duidelijke splitsing in homologe chromosomen.
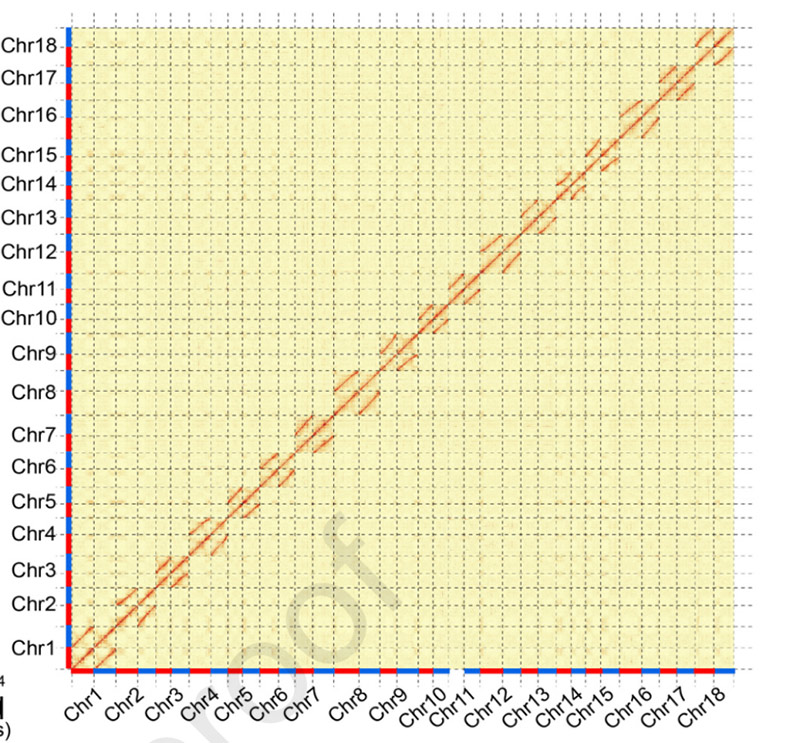
Hu W et al.,Moleculaire plant, 2021
4.Hi-C heatmap op genoomassemblage van twee Ficus-soorten:F.microcarpa(verankeringsgraad: 99,3%) enF.hispida (verankeringsratio: 99,7%)
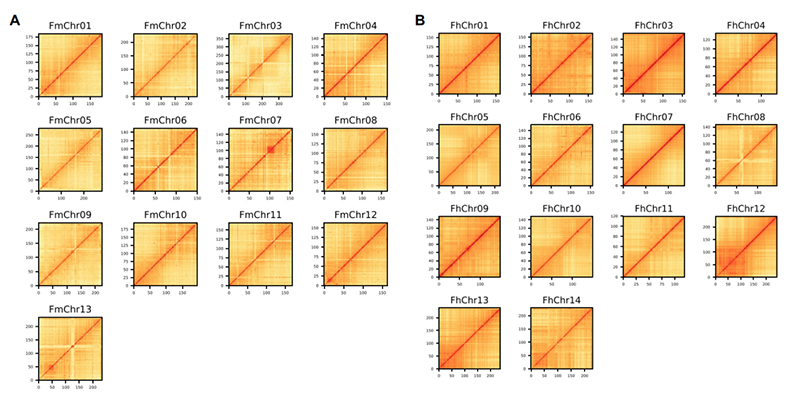
Zhang X et al.,Cel, 2020
BMK-zaak
Genomen van de banyanboom en de bestuiverswesp bieden inzicht in de co-evolutie van de vijgenwesp
Gepubliceerd: Cel, 2020
Sequentiestrategie:
F. microcarpa genoom: Ca.84 X PacBio RSII (36,87 GB) + Hi-C (44 GB)
F. hispidagenoom: Ca.97 X PacBio RSII (36,12 GB) + Hi-C (60 GB)
Eupristina verticillatagenoom: Ca.170 X PacBio RSII (65 GB)
Belangrijkste resultaten
1. Twee banyanboomgenomen en één bestuiverwespengenoom werden geconstrueerd met behulp van PacBio-sequencing, Hi-C en koppelingskaart.
(1)F. microcarpagenoom: Er werd een samenstel van 426 Mb (97,7% van de geschatte genoomgrootte) tot stand gebracht met een contig N50 van 908 Kb, een BUSCO-score van 95,6%.In totaal werden 423 Mb-sequenties door Hi-C aan 13 chromosomen verankerd.Genoomannotatie leverde 29.416 eiwitcoderende genen op.
(2)F. Hispidagenoom: Een samenstel van 360 Mb (97,3% van de geschatte genoomgrootte) werd opgeleverd met een contig N50 van 492 Kb en een BUSCO-score van 97,4%.Een totaal van 359 Mb-sequenties werd door Hi-C op 14 chromosomen verankerd en was in hoge mate identiek aan de koppelingskaart met hoge dichtheid.
(3)Eupristina verticillatagenoom: Er werd een samenstel van 387 Mb (geschatte genoomgrootte: 382 Mb) tot stand gebracht met een contig N50 van 3,1 Mb en een BUSCO-score van 97,7%.
2.Vergelijkende genomica-analyse onthulde een groot aantal structuurvariaties tussen tweeFicusgenomen, die een onschatbare genetische bron vormden voor adaptieve evolutiestudies.Deze studie verschafte voor het eerst inzicht in de co-evolutie van vijgenwesp op genomisch niveau.
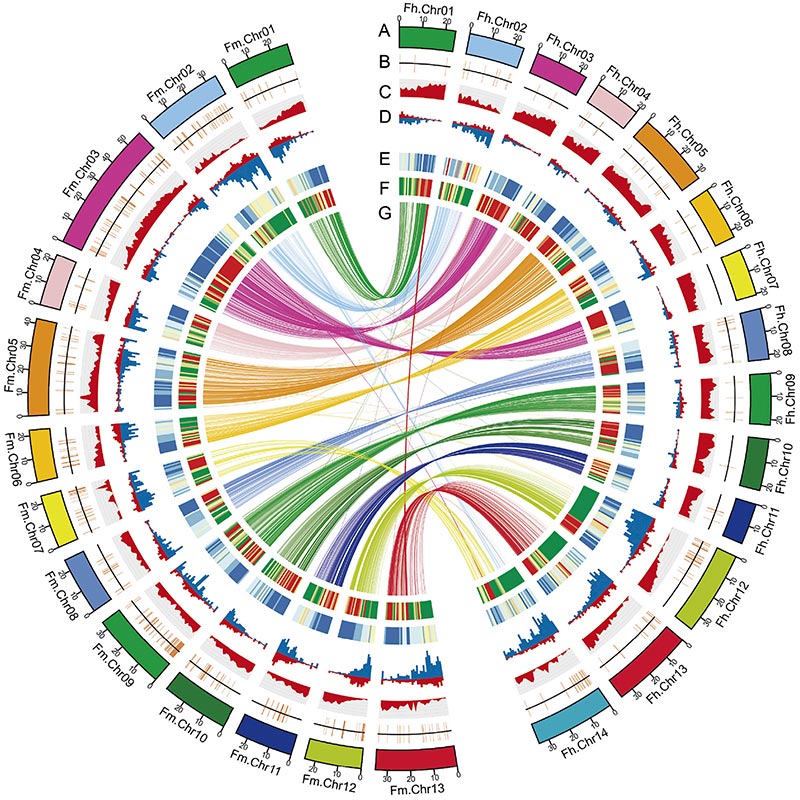 Circosdiagram over genomische kenmerken van tweeFicusgenomen, inclusief chromosomen, segmentale duplicaties (SD's), transposons (LTR, TE's, DNA TE's), genexpressie en syntenie | 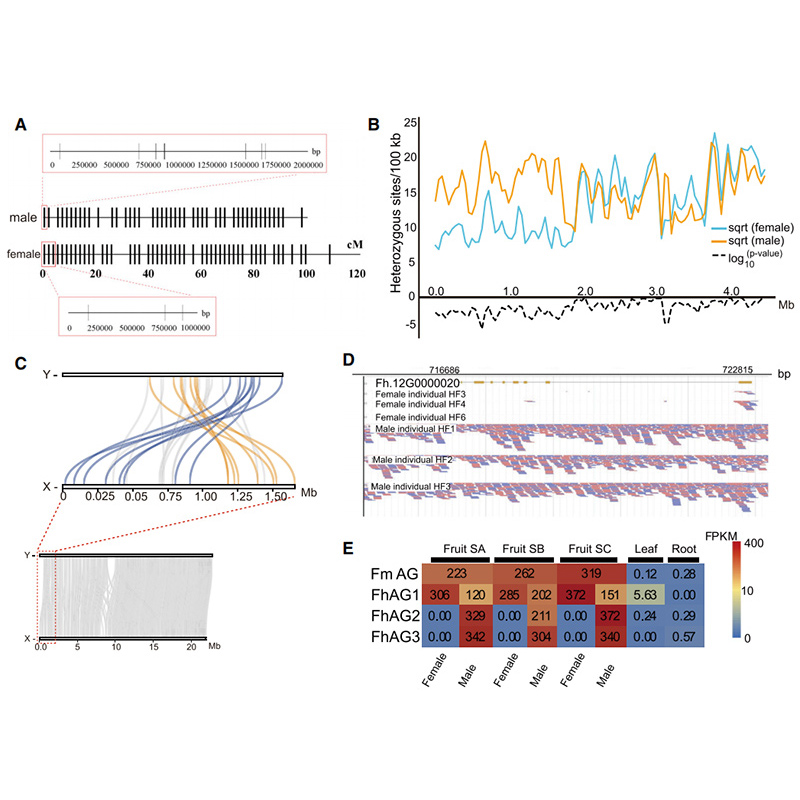 Identificatie van het Y-chromosoom en kandidaatgen voor geslachtsbepaling |
Zhang, X., et al."Genomen van de banyanboom en bestuiverwesp bieden inzicht in de co-evolutie van vijgenwesp."Cel 183.4(2020).





