

Bulked Segregant-analyse






Servicevoordelen
Takagi et al., Het plantentijdschrift, 2013
● Nauwkeurige lokalisatie: bulks mengen met 30+30 tot 200+200 personen om achtergrondgeluiden te minimaliseren;niet-synonieme, op mutatanten gebaseerde voorspelling van kandidaatregio's.
● Uitgebreide analyse: diepgaande annotatie van kandidaatgenfuncties, waaronder NR, SwissProt, GO, KEGG, COG, KOG, enz.
● Snellere doorlooptijd: Snelle genlokalisatie binnen 45 werkdagen.
● Uitgebreide ervaring: BMK heeft bijgedragen aan de lokalisatie van duizenden eigenschappen, waaronder diverse soorten zoals gewassen, aquatische producten, bossen, bloemen, fruit, enz.
Servicespecificaties
Bevolking:
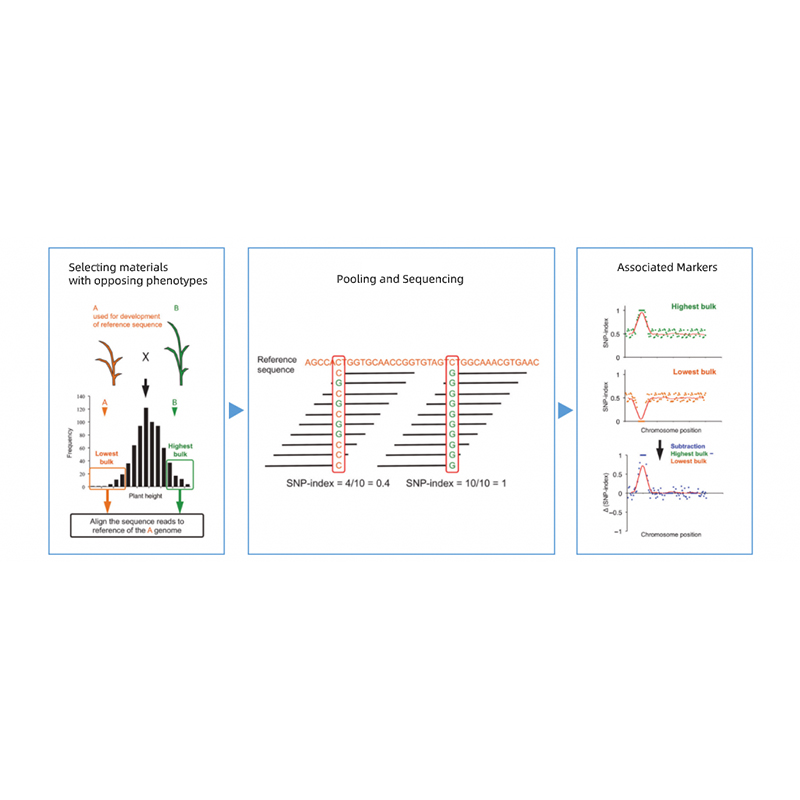
Het scheiden van nakomelingen van ouders met tegengestelde fenotypes.
bijv. F2-nakomelingen, terugkruising (BC), recombinante inteeltlijn (RIL)
Mengzwembad
Voor kwalitatieve kenmerken: 30 tot 50 individuen (minimaal 20)/bulk
Voor kwantitatieve tratis: top 5% tot 10% individuen met extreme fenotypes in de hele populatie (minimaal 30+30).
Aanbevolen sequentiediepte
Tenminste 20X/ouder en 1X/nakomeling individu (bijvoorbeeld voor een mengpool van 30+30 nakomelingen zal de sequentiediepte 30X per bulk zijn)
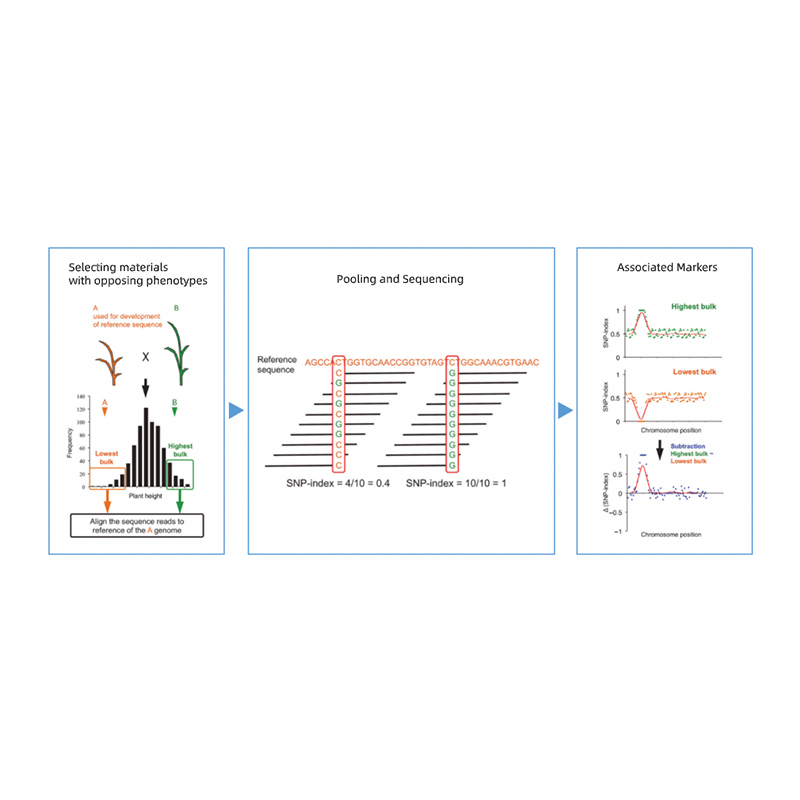
Bio-informatica analyses
● Hersequencing van het hele genoom
● Gegevensverwerking
● SNP/Indel-oproepen
● Screening van kandidaatregio's
● Annotatie van de kandidaat-genfunctie
Monstervereisten en levering
Voorbeeldvereisten:
Nucleotiden:
| gDNA-monster | Weefselmonster |
| Concentratie: ≥30 ng/μl | Planten: 1-2 g |
| Hoeveelheid: ≥2 μg (volume ≥15 μl) | Dieren: 0,5-1 g |
| Zuiverheid: OD260/280= 1,6-2,5 | Volbloed: 1,5 ml |
Servicewerkstroom

Experimentontwerp

Levering van monsters

RNA-extractie

Bouw van bibliotheek

Volgorde aanbrengen in

Gegevensanalyse

After-sales diensten
1. Associatieanalyse gebaseerd op Euclidische afstand (ED) om kandidaatregio te identificeren.In de volgende figuur
X-as: chromosoomnummer;Elke stip vertegenwoordigt een ED-waarde van een SNP.De zwarte lijn komt overeen met de aangepaste ED-waarde.Een hogere ED-waarde duidt op een significanter verband tussen de site en het fenotype.Een rode stippellijn geeft de drempel van significante associatie weer.
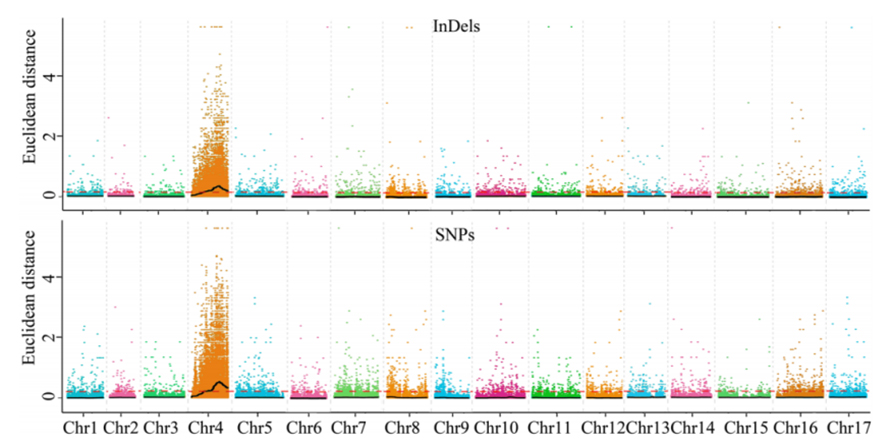
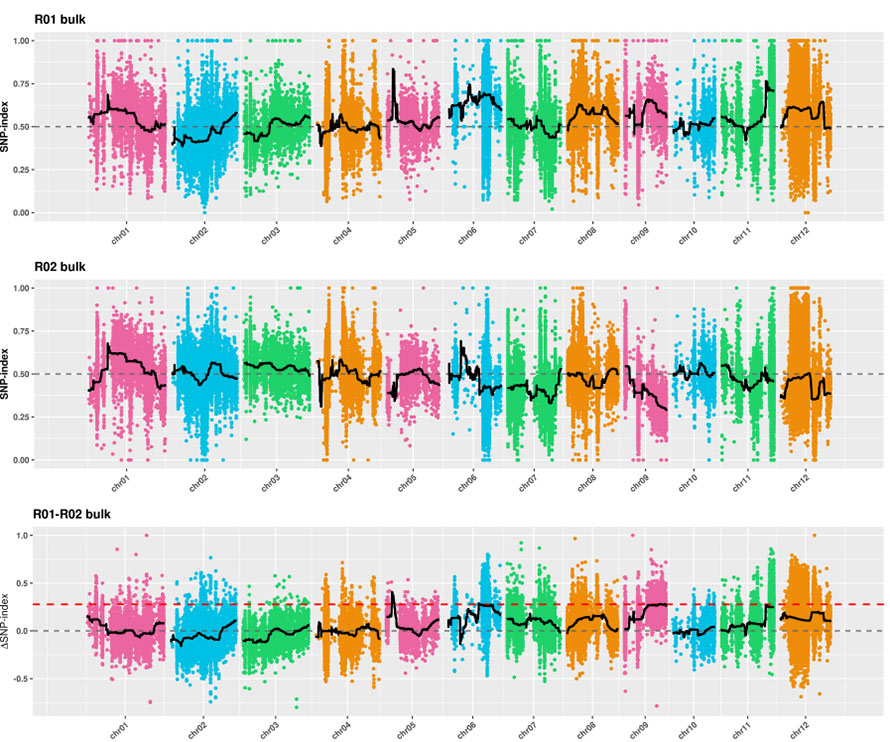
2. Associatieanalyse gebaseerd op geen SNP-index
X-as: chromosoomnummer;Elke stip vertegenwoordigt de SNP-indexwaarde.De zwarte lijn staat voor de gepaste SNP-indexwaarde.Hoe groter de waarde, hoe significanter de associatie.
BMK-zaak
De kwantitatieve kenmerklocus Fnl7.1 met het belangrijkste effect codeert voor een overvloedig eiwit in de late embryogenese dat geassocieerd is met de lengte van de vruchthals in komkommer
Gepubliceerd: Tijdschrift voor plantenbiotechnologie, 2020
Sequentiestrategie:
Ouders (Jin5-508, YN): Herschikking van het hele genoom voor 34× en 20×.
DNA-pools (50 met lange hals en 50 met korte hals): Herschikking voor 61× en 52×
Belangrijkste resultaten
In deze studie werd een gescheiden populatie (F2 en F2:3) gegenereerd door de komkommerlijn Jin5-508 met lange nek en YN met korte nek te kruisen.Er werden twee DNA-pools samengesteld door 50 individuen met extreem lange nek en 50 individuen met extreem korte nek.QTL met groot effect werd op Chr07 geïdentificeerd door BSA-analyse en traditionele QTL-mapping.Het kandidaatgebied werd verder verkleind door middel van nauwkeurige mapping, kwantificering van genexpressie en transgene experimenten, die het sleutelgen onthulden bij het controleren van de neklengte, CsFnl7.1.Bovendien bleek polymorfisme in het CsFnl7.1-promotergebied geassocieerd te zijn met overeenkomstige expressie.Verdere fylogenetische analyse suggereerde dat de Fnl7.1-locus zeer waarschijnlijk afkomstig is uit India.
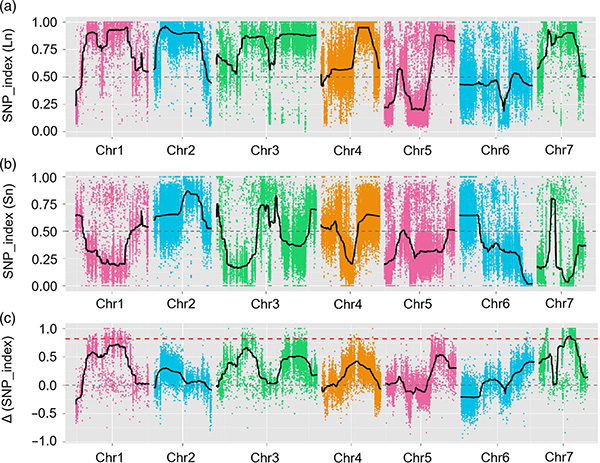 QTL-mapping in BSA-analyse om kandidaat-regio geassocieerd met komkommerhalslengte te identificeren | 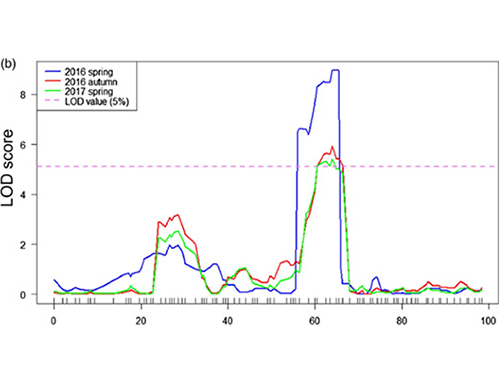 LOD-profielen van QTL met neklengte van komkommer geïdentificeerd op Chr07 |
Xu, X., et al."De kwantitatieve eigenschapslocus Fnl7.1 met het grootste effect codeert voor een overvloedig eiwit in de late embryogenese dat geassocieerd is met de lengte van de vruchthals in komkommer."Plantenbiotechnologie Journal 18.7 (2020).





