

Specifiska lokusa pastiprinātā fragmentu secība (SLAF-Seq)





Sīkāka informācija par pakalpojumu
Tehniskā shēma
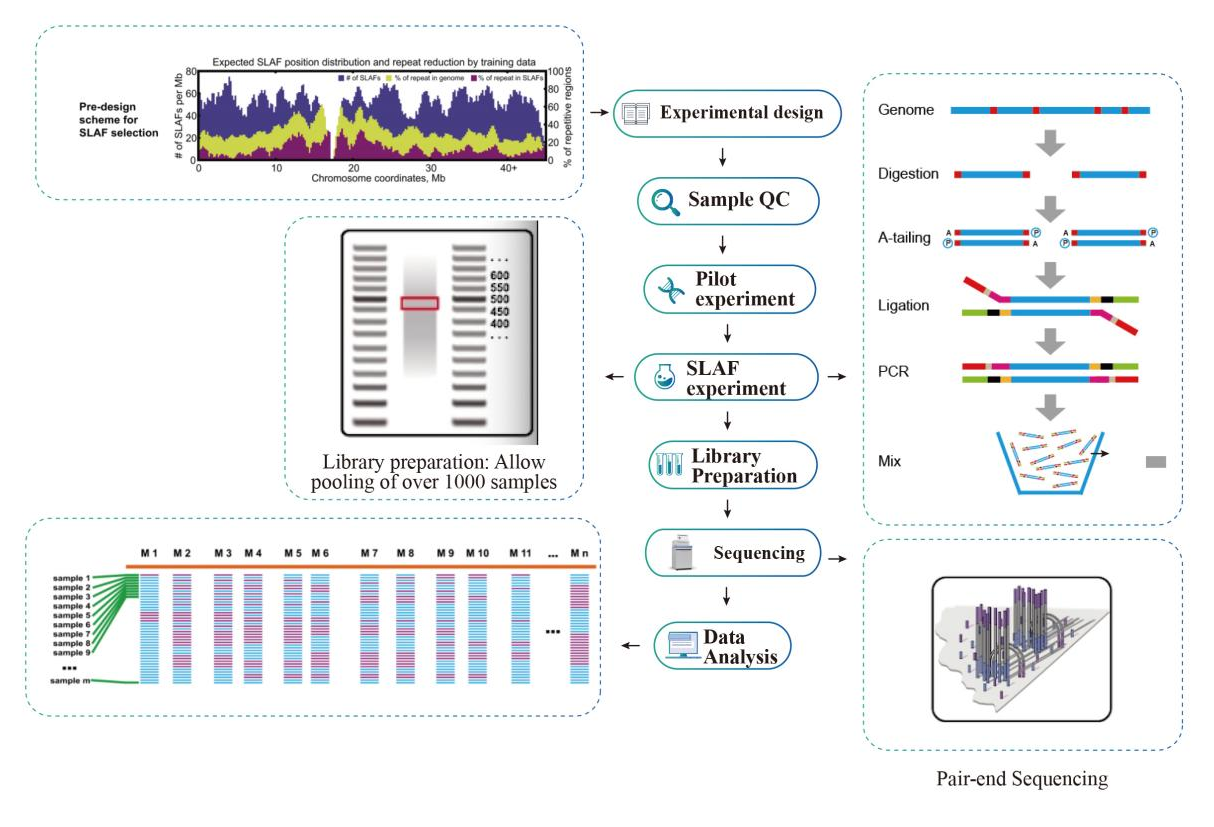
Darba plūsma

Pakalpojuma priekšrocības
Augsta marķieru atklāšanas efektivitāte- Augstas caurlaidības sekvencēšanas tehnoloģija palīdz SLAF-Seq atklāt simtiem tūkstošu tagu visā genomā.
Zema atkarība no genoma- To var attiecināt uz sugām ar vai bez atsauces genoma.
Elastīgs shēmu dizains- Viena enzīma, divu enzīmu, vairāku enzīmu gremošanas un dažāda veida enzīmus var atlasīt, lai apmierinātu dažādus pētniecības mērķus vai sugas.Iepriekšēja novērtēšana in silico tiek izmantota, lai nodrošinātu optimālu enzīmu dizainu.
Efektīva fermentatīvā gremošana- Iepriekšējais eksperiments tika veikts, lai optimizētu apstākļus, kas padara formālo eksperimentu stabilu un uzticamu.Fragmentu savākšanas efektivitāte var sasniegt vairāk nekā 95%.
Vienmērīgi sadalīti SLAF tagi- SLAF tagi ir vienmērīgi sadalīti visās hromosomās vislielākajā mērā, sasniedzot vidēji 1 SLAF uz 4 kb.
Efektīva atkārtošanās novēršana- Atkārtota secība SLAF-Seq datos ir samazināta līdz zemākai par 5%, īpaši sugās ar augstu atkārtošanās līmeni, piemēram, kviešiem, kukurūzai utt.
Plaša pieredze- Vairāk nekā 2000 slēgtu SLAF-Seq projektu simtiem sugu, kas aptver augus, zīdītājus, putnus, kukaiņus, ūdensorganismus utt.
Pašu izstrādāta bioinformātiskā darbplūsma- BMKGENE izstrādāja integrētu bioinformātisko darbplūsmu SLAF-Seq, lai nodrošinātu galīgās produkcijas uzticamību un precizitāti.
Pakalpojuma specifikācijas
| Platforma | Konc. (ng/gl) | Kopā (ug) | OD260/280 |
| Illumina NovaSeq | >35 | >1.6(Sējums>15μl) | 1,6-2,5 |
Ieteicamā secības noteikšanas stratēģija
Sekvences dziļums: 10X/Tag
| Genoma izmērs | Ieteicamie SLAF tagi |
| < 500 Mb | 100 K vai WGS |
| 500 Mb–1 Gb | 100 K |
| 1 Gb - 2 Gb | 200 K |
| Milzu vai sarežģīti genomi | 300–400 000 |
| Lietojumprogrammas
| Ieteicams Iedzīvotāju skala
| Sekvences stratēģija un dziļums
| |
| Dziļums
| Atzīmes numurs
| ||
| GWAS
| Parauga numurs ≥ 200
| 10X
|
Saskaņā ar genoma lielums
|
| Ģenētiskā evolūcija
| Katra atsevišķas personas apakšgrupa ≥ 10; kopējie paraugi ≥ 30
| 10X
| |
Ieteicamā paraugu piegāde
Tvertne: 2 ml centrifūgas caurule
Lielākajai daļai paraugu mēs iesakām neglabāt etanolā.
Paraugu marķējums: paraugiem jābūt skaidri marķētiem un identiskiem ar iesniegto parauga informācijas veidlapu.
Sūtījums: Sausais ledus: Paraugi vispirms jāiepako maisos un jāierok sausajā ledū.
Pakalpojuma darbplūsma






QC paraugs
Piloteksperiments
SLAF-eksperiments
Bibliotēkas sagatavošana
Secība
Datu analīze
Pēcpārdošanas pakalpojumi
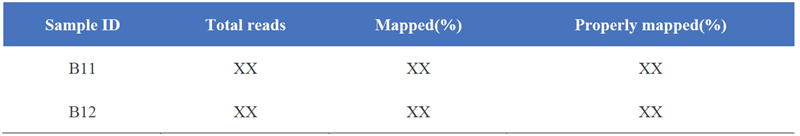
1. Kartes rezultātu statistika

2. SLAF marķieru izstrāde

3. Variāciju anotācija

| gads | Žurnāls | IF | Nosaukums | Lietojumprogrammas |
| 2022. gads | Dabas komunikācijas | 17.694 | Koka peonijas giga-hromosomu un giga-genoma genomiskais pamats Paeonia ostii | SLAF-GWAS |
| 2015. gads | Jaunais fitologs | 7.433 | Domestācijas pēdas noenkuro agronomiski nozīmīgus genoma reģionus sojas pupiņas | SLAF-GWAS |
| 2022. gads | Uzlaboto pētījumu žurnāls | 12.822 | Gossypium barbadense mākslīgās introgresijas visā genomā G. hirsutum atklāj izcilus lokusus, lai vienlaikus uzlabotu kokvilnas šķiedras kvalitāti un ražu iezīmes | SLAF-Evolūcijas ģenētika |
| 2019. gads | Molekulārais augs | 10.81 | Populācijas genomiskā analīze un De Novo asambleja atklāj Weedy izcelsmi Rīsi kā evolūcijas spēle | SLAF-Evolūcijas ģenētika |
| 2019. gads | Dabas ģenētika | 31.616 | Parastās karpas Cyprinus carpio genoma secība un ģenētiskā daudzveidība | SLAF-Saiknes karte |
| 2014. gads | Dabas ģenētika | 25.455 | Kultivēto zemesriekstu genoms sniedz ieskatu pākšaugu kariotipos, poliploīdos evolūcija un labības pieradināšana. | SLAF-Saiknes karte |
| 2022. gads | Augu biotehnoloģijas žurnāls | 9.803 | ST1 identifikācija atklāj atlasi, kas ietver sēklu morfoloģijas autostopu un eļļas saturs sojas pupu pieradināšanas laikā | SLAF-Markeru izstrāde |
| 2022. gads | Starptautiskais molekulāro zinātņu žurnāls | 6.208 | Kviešu-Leymus mollis 2Ns (2D) identifikācija un DNS marķiera izstrāde Disomiskā hromosomu aizstāšana | SLAF-Markeru izstrāde |





