

Augstas caurlaidības genotipēšana, īpaši liela mēroga populācijā, ir būtisks solis ģenētisko asociāciju pētījumos, kas nodrošina ģenētisko pamatu funkcionālu gēnu atklāšanai, evolūcijas analīzei utt. Tā vietā, lai veiktu dziļu visa genoma atkārtotu sekvencēšanu, samazināta reprezentācijas genoma sekvencēšana (RRGS). ) tiek ieviests, lai samazinātu sekvencēšanas izmaksas vienam paraugam, vienlaikus saglabājot saprātīgu efektivitāti ģenētisko marķieru atklāšanā.To parasti panāk, ekstrahējot restrikcijas fragmentu noteiktā lieluma diapazonā, ko sauc par samazinātu reprezentācijas bibliotēku (RRL).Specifiska lokusa pastiprināta fragmentu sekvencēšana (SLAF-Seq) ir pašu izstrādāta stratēģija de novo SNP atklāšanai un lielu populāciju SNP genotipēšanai.
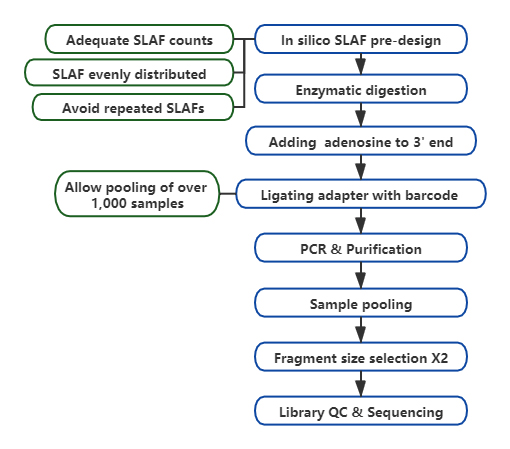
Tehniskā darba plūsma
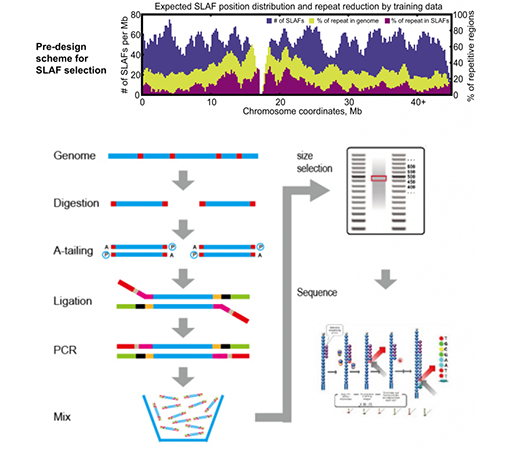
SLAF pret esošajām RRL metodēm
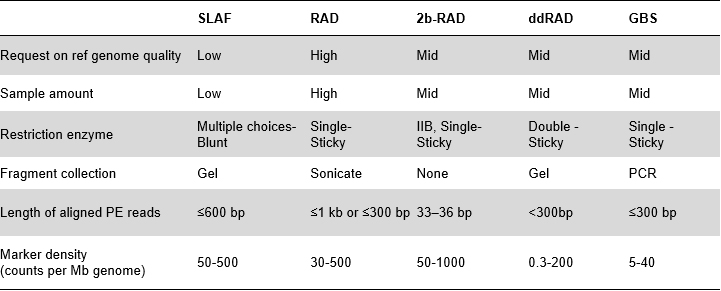
SLAF priekšrocības
Augstāka ģenētisko marķieru atklāšanas efektivitāte– Apvienojumā ar augstas caurlaidības sekvencēšanas tehnoloģiju, SLAF-Seq varētu iegūt simtiem tūkstošu tagu, kas atklāti visā genomā, lai izpildītu dažādu pētniecības projektu pieprasījumu ar vai bez atsauces genoma.
Pielāgots un elastīgs eksperimentāls dizains– Dažādiem pētniecības mērķiem vai sugām ir pieejamas dažādas fermentatīvās gremošanas stratēģijas, tostarp viena enzīma, divu enzīmu un vairāku enzīmu gremošanas stratēģijas.Gremošanas stratēģija tiks iepriekš novērtēta in silico, lai nodrošinātu optimālu enzīmu dizainu.
Augsta fermentatīvās gremošanas efektivitāte– Iepriekš izstrādāta fermentatīvā gremošana nodrošina vienmērīgāku SLAF sadalījumu hromosomā.Fragmentu savākšanas efektivitāte var sasniegt vairāk nekā 95%.
Izvairieties no atkārtotas secības– Atkārtotu secību procentuālā daļa SLAF-Seq datos ir samazināta līdz mazākai par 5%, īpaši sugās ar augstu atkārtojošos elementu līmeni, piemēram, kviešiem, kukurūzai utt.
Pašu izstrādāta bioinformātiskā darbplūsma– BMK izstrādāja integrētu bioinformātisko darbplūsmu, kas piemērojama SLAF-Seq tehnoloģijai, lai nodrošinātu gala izvades uzticamību un precizitāti.
SLAF pielietojums
Ģenētiskās saiknes karte
Augsta blīvuma ģenētiskās kartes uzbūve un lokusu identificēšana, kas kontrolē krizantēmas (Chrysanthemum x morifolium Ramat.) ziedu tipa pazīmes.
Žurnāls: Dārzkopības pētījumi Publicēts: 2020.7.2020

GWAS
Kandidāta gēna identificēšana, kas saistīta ar izofavona saturu sojas pupu sēklās, izmantojot genoma mēroga asociāciju un saiknes kartēšanu
Žurnāls: The Plant Journal Publicēts: 2020.08.2020

Evolūcijas ģenētika
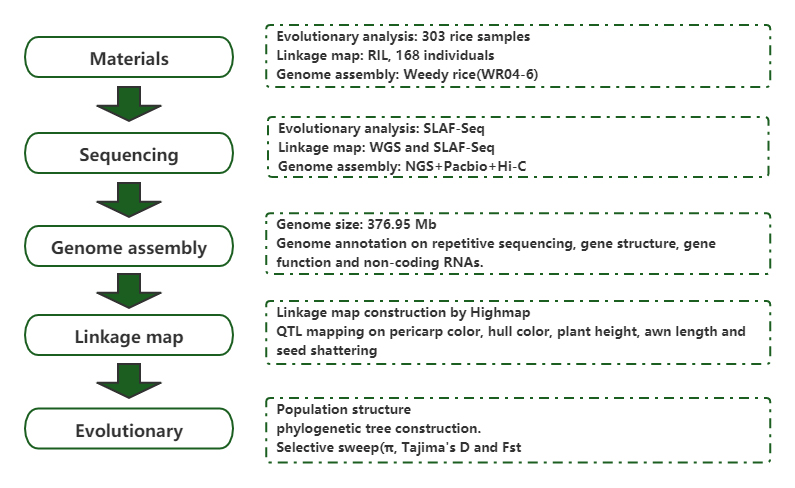
Populācijas genoma analīze un de novo montāža atklāj nezāļu rīsu kā evolūcijas spēles izcelsmi
Žurnāls: Molecular Plant Publicēts: 2019.5.2019
Lielapjoma segregācijas analīze (BSA)
GmST1, kas kodē sulfotransferāzi, nodrošina rezistenci pret sojas pupu mozaīkas vīrusa celmiem G2 un G3
Žurnāls: Plant, Cell&Environment Publicēts: 2021.04.2021

Atsauce
Sun X, Liu D, Zhang X u.c.SLAF-Seq: efektīva liela mēroga de novo SNP atklāšanas un genotipa noteikšanas metode, izmantojot augstas caurlaidības sekvencēšanu [J].Plos one, 2013, 8(3):e58700
Dziesma X, Xu Y, Gao K u.c.Augsta blīvuma ģenētiskās kartes uzbūve un lokusu identificēšana, kas kontrolē ziedu tipa pazīmes krizantēmā (Chrysanthemum × morifolium Ramat.).Hortic Res.2020; 7:108.
Wu D, Li D, Zhao X u.c.Kandidāta gēna, kas saistīts ar izoflavona saturu sojas pupu sēklās, identificēšana, izmantojot genoma mēroga asociāciju un saiknes kartēšanu.Plant J. 2020;104(4): 950-963.
Sun J, Ma D, Tang L u.c.Populācijas genomiskā analīze un De Novo asambleja atklāj nezāļu rīsu kā evolūcijas spēles izcelsmi.Molu rūpnīca.2019;12(5):632-647.Molu rūpnīca.2018. gads;11(11):1360-1376.
Zhao X, Jing Y, Luo Z u.c.GmST1, kas kodē sulfotransferāzi, nodrošina rezistenci pret sojas pupu mozaīkas vīrusa celmiem G2 un G3.Augu šūnu vide.2021;10.1111/gab.14066
Izlikšanas laiks: Jan-04-2022