

ການຈັດລຳດັບ Fragment Amplified Specific-Locus (SLAF-Seq)





ລາຍລະອຽດການບໍລິການ
ໂຄງການທາງວິຊາການ
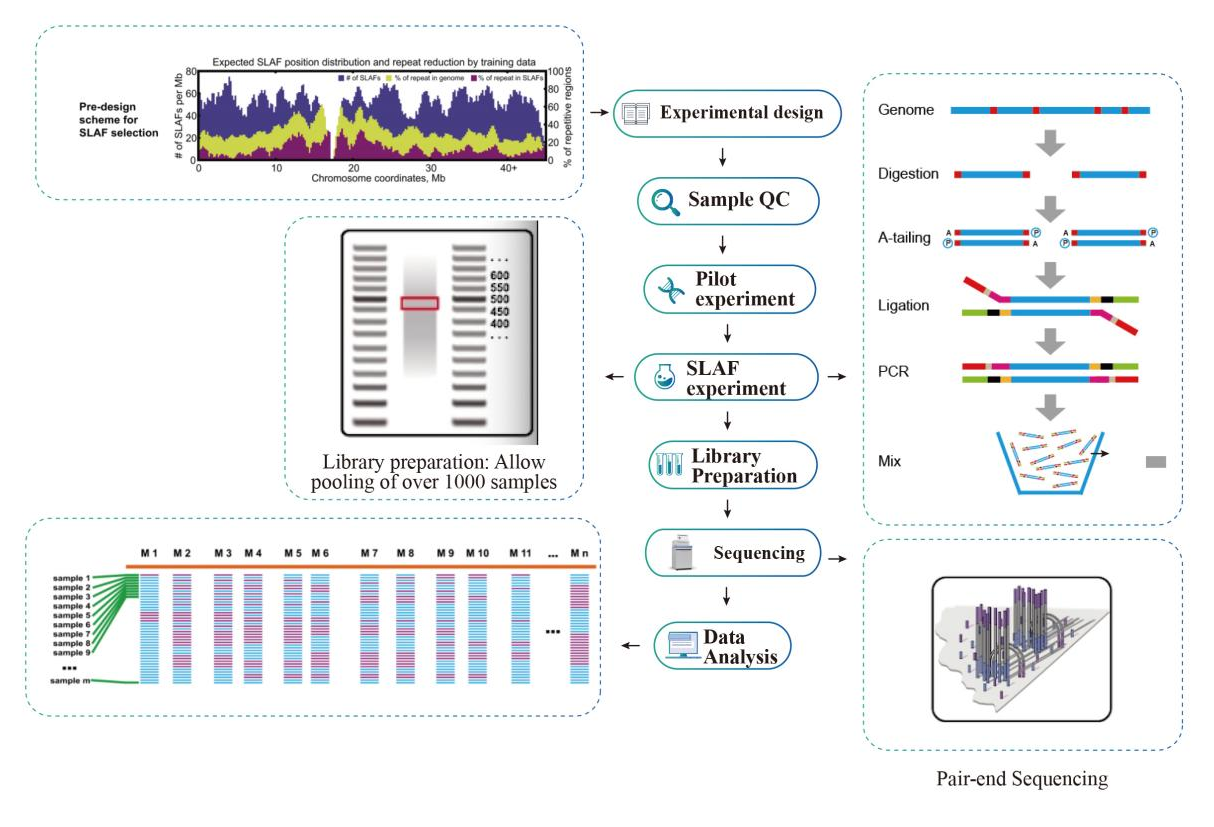
ກະແສວຽກ

ຂໍ້ໄດ້ປຽບການບໍລິການ
ປະສິດທິພາບການຄົ້ນພົບເຄື່ອງໝາຍສູງ- ເທກໂນໂລຍີການຈັດລໍາດັບທີ່ຜ່ານຄວາມໄວສູງຊ່ວຍ SLAF-Seq ໃນການຄົ້ນພົບຫຼາຍຮ້ອຍພັນແທັກພາຍໃນ genome ທັງຫມົດ.
ການເອື່ອຍອີງຕ່ໍາສຸດຂອງ genome- ມັນສາມາດຖືກນໍາໃຊ້ກັບຊະນິດທີ່ມີຫຼືບໍ່ມີ genome ກະສານອ້າງອີງ.
ການອອກແບບໂຄງການທີ່ມີຄວາມຍືດຫຍຸ່ນ- ເອນໄຊດຽວ, ສອງເອນໄຊ, ການຍ່ອຍອາຫານຫຼາຍອັນ ແລະເອນໄຊຊະນິດຕ່າງໆ, ທັງໝົດສາມາດເລືອກໄດ້ເພື່ອຕອບສະໜອງເປົ້າໝາຍການຄົ້ນຄວ້າ ຫຼືຊະນິດຕ່າງໆ.ການປະເມີນເບື້ອງຕົ້ນໃນຊິລິໂຄແມ່ນໃຊ້ເພື່ອຮັບປະກັນການອອກແບບ enzyme ທີ່ດີທີ່ສຸດ.
ການຍ່ອຍອາຫານ enzymatic ປະສິດທິພາບ- ການທົດລອງທາງສ່ວນຫນ້າໄດ້ຖືກປະຕິບັດເພື່ອເພີ່ມປະສິດທິພາບເງື່ອນໄຂ, ເຊິ່ງເຮັດໃຫ້ການທົດລອງຢ່າງເປັນທາງການມີຄວາມຫມັ້ນຄົງແລະເຊື່ອຖືໄດ້.ປະສິດທິພາບການເກັບກໍາ Fragment ສາມາດບັນລຸໄດ້ຫຼາຍກວ່າ 95%.
tags SLAF ແຈກຢາຍຢ່າງເທົ່າທຽມກັນ- tags SLAF ຖືກແຈກຢາຍຢ່າງເທົ່າທຽມກັນໃນທຸກໂຄໂມໂຊມໃນລະດັບສູງສຸດ, ບັນລຸໄດ້ໂດຍສະເລ່ຍ 1 SLAF ຕໍ່ 4 kb.
ການຫຼີກລ່ຽງການຊໍ້າຄືນຢ່າງມີປະສິດທິພາບ- ລໍາດັບການຊໍ້າຄືນໃນຂໍ້ມູນ SLAF-Seq ແມ່ນຫຼຸດລົງຕໍ່າກວ່າ 5%, ໂດຍສະເພາະໃນຊະນິດທີ່ມີລະດັບການຊໍ້າຄືນສູງເຊັ່ນ: ເຂົ້າສາລີ, ສາລີ, ແລະອື່ນໆ.
ປະສົບການຢ່າງກວ້າງຂວາງ- ຫຼາຍກວ່າ 2000 ໂຄງການປິດ SLAF-Seq ກ່ຽວກັບຫຼາຍຮ້ອຍຊະນິດທີ່ກວມເອົາພືດ, ສັດລ້ຽງລູກດ້ວຍນົມ, ນົກ, ແມງໄມ້, ສັດນ້ໍາ, ແລະອື່ນໆ.
ຂະບວນການເຮັດວຽກ bioinformatic ພັດທະນາຕົນເອງ- ຂະບວນການເຮັດວຽກທາງດ້ານຊີວະວິທະຍາແບບປະສົມປະສານສໍາລັບ SLAF-Seq ໄດ້ຖືກພັດທະນາໂດຍ BMKGENE ເພື່ອຮັບປະກັນຄວາມຫນ້າເຊື່ອຖືແລະຄວາມຖືກຕ້ອງຂອງຜົນຜະລິດສຸດທ້າຍ.
ຂໍ້ມູນຈໍາເພາະການບໍລິການ
| ເວທີ | Conc.(ng/gl) | ທັງໝົດ (ug) | OD260/280 |
| Illumina NovaSeq | >35 | >1.6(ເຫຼັ້ມ > 15μl) | 1.6-2.5 |
ຍຸດທະສາດການຈັດລໍາດັບທີ່ແນະນໍາ
ຄວາມເລິກຂອງລໍາດັບ: 10X/Tag
| ຂະໜາດພັນທຸ ກຳ | ແນະນຳປ້າຍ SLAF |
| < 500 Mb | 100K ຫຼື WGS |
| 500 Mb- 1 Gb | 100 K |
| 1 Gb -2 Gb | 200 K |
| genomes ຍັກໃຫຍ່ຫຼືສະລັບສັບຊ້ອນ | 300 - 400K |
| ຄໍາຮ້ອງສະຫມັກ
| ແນະນຳ ຂະໜາດປະຊາກອນ
| ການຈັດລໍາດັບຍຸດທະສາດແລະຄວາມເລິກ
| |
| ຄວາມເລິກ
| ໝາຍເລກແທັກ
| ||
| GWAS
| ຈໍານວນຕົວຢ່າງ ≥ 200
| 10X
|
ອີງຕາມ ຂະຫນາດຂອງ genome
|
| ວິວັດທະນາການທາງພັນທຸກໍາ
| ບຸກຄົນຂອງແຕ່ລະຄົນ ກຸ່ມຍ່ອຍ ≥ 10; ຕົວຢ່າງທັງໝົດ≥ 30
| 10X
| |
ການຈັດສົ່ງຕົວຢ່າງທີ່ແນະນໍາ
ບັນຈຸ: ທໍ່ centrifuge 2 ml
ສໍາລັບຕົວຢ່າງສ່ວນໃຫຍ່, ພວກເຮົາແນະນໍາໃຫ້ບໍ່ເກັບຮັກສາໃນເອທານອນ.
ການຕິດສະຫຼາກຕົວຢ່າງ: ຕົວຢ່າງຕ້ອງມີການຕິດສະຫຼາກຢ່າງຊັດເຈນ ແລະຄືກັນກັບແບບຟອມຂໍ້ມູນຕົວຢ່າງທີ່ສົ່ງມາ.
ການຂົນສົ່ງ: ນໍ້າກ້ອນແຫ້ງ: ຕົວຢ່າງຕ້ອງຖືກບັນຈຸໃສ່ຖົງກ່ອນ ແລະຝັງໄວ້ໃນນໍ້າກ້ອນແຫ້ງ.
ຂະບວນການບໍລິການ






ຕົວຢ່າງ QC
ການທົດລອງທົດລອງ
SLAF-ທົດລອງ
ການກະກຽມຫ້ອງສະຫມຸດ
ການຈັດລໍາດັບ
ການວິເຄາະຂໍ້ມູນ
ບໍລິການຫຼັງການຂາຍ
1. ສະຖິຕິຜົນແຜນທີ່

2. ການພັດທະນາເຄື່ອງໝາຍ SLAF

3. ຄໍາບັນຍາຍການປ່ຽນແປງ

| ປີ | ວາລະສານ | IF | ຫົວຂໍ້ | ຄໍາຮ້ອງສະຫມັກ |
| 2022 | ການສື່ສານທໍາມະຊາດ | 17.694 | ພື້ນຖານພັນທຸກໍາຂອງ giga-chromosomes ແລະ giga-genome ຂອງ peony ຕົ້ນໄມ້ Paeonia ostii | SLAF-GWAS |
| 2015 | ແພດສາດໃໝ່ | 7.433 | ຮອຍຕີນຂອງພາຍໃນປະເທດສະມໍເຂດພັນທຸກໍາທີ່ມີຄວາມສໍາຄັນທາງດ້ານກະສິກໍາໃນ ຖົ່ວເຫຼືອງ | SLAF-GWAS |
| 2022 | ວາລະສານການຄົ້ນຄວ້າຂັ້ນສູງ | 12.822 | Genome-wide introgressions ທຽມຂອງ Gossypium barbadense ເຂົ້າໄປໃນ G. hirsutum ເປີດເຜີຍຈຸດທີ່ເໜືອກວ່າສຳລັບການປັບປຸງຄຸນນະພາບຂອງເສັ້ນໃຍຝ້າຍພ້ອມໆກັນ ແລະໃຫ້ຜົນຜະລິດ ລັກສະນະ | SLAF-Evolutionary ພັນທຸ ກຳ |
| 2019 | ພືດໂມເລກຸນ | 10.81 | ການວິເຄາະ Genomic ປະຊາກອນແລະກອງປະຊຸມ De Novo ເປີດເຜີຍຕົ້ນກໍາເນີດຂອງ Weedy ເຂົ້າເປັນເກມວິວັດທະນາການ | SLAF-Evolutionary ພັນທຸ ກຳ |
| 2019 | ພັນທຸ ກຳ ທໍາມະຊາດ | 31.616 | ລໍາດັບພັນທຸກໍາແລະຄວາມຫຼາກຫຼາຍທາງພັນທຸກໍາຂອງປາພືດທົ່ວໄປ, Cyprinus carpio | ແຜນທີ່ການເຊື່ອມໂຍງ SLAF |
| 2014 | ພັນທຸ ກຳ ທໍາມະຊາດ | 25.455 | genome ຂອງຖົ່ວດິນທີ່ປູກຝັງໃຫ້ຄວາມເຂົ້າໃຈກ່ຽວກັບ karyotypes legume, polyploid ວິວັດທະນາການ ແລະ ການປູກພືດພາຍໃນປະເທດ. | ແຜນທີ່ການເຊື່ອມໂຍງ SLAF |
| 2022 | ວາລະສານເຕັກໂນໂລຊີຊີວະພາບພືດ | 9.803 | ການກໍານົດຕົວຂອງ ST1 ສະແດງໃຫ້ເຫັນເຖິງການຄັດເລືອກທີ່ກ່ຽວຂ້ອງກັບການຍ່າງປ່າຂອງເມັດພືດ ແລະປະລິມານນ້ໍາມັນໃນໄລຍະການຜະລິດຖົ່ວເຫຼືອງ | ການພັດທະນາ SLAF-Marker |
| 2022 | ວາລະສານສາກົນຂອງວິທະຍາສາດໂມເລກຸນ | 6.208 | ການລະບຸຕົວຕົນ ແລະການພັດທະນາເຄື່ອງໝາຍ DNA ສໍາລັບ Wheat-Leymus mollis 2Ns (2D) ການທົດແທນ Chromosome Disomic | ການພັດທະນາ SLAF-Marker |





