

High-Throughput Genotyping, besonnesch op grousser Bevëlkerung, ass e fundamentale Schrëtt an der genetescher Associatiounsstudien, déi genetesch Basis fir funktionell Genentdeckung, evolutiv Analyse, etc. ) gëtt agefouert fir d'Sequenzéierungskäschte pro Probe ze minimiséieren, wärend raisonnabel Effizienz bei der Genetescher Marker Entdeckung erhalen.Dëst gëtt allgemeng erreecht andeems d'Restriktiounsfragment an engem bestëmmte Gréisstberäich extrahéiert gëtt, wat reduzéiert Representatiounsbibliothéik (RRL) genannt gëtt.Spezifesch-Locus amplifizéiert Fragmenter Sequencing (SLAF-Seq) ass eng selbstentwéckelt Strategie fir de novo SNP Entdeckung an SNP Genotyping vu grousse Populatiounen.
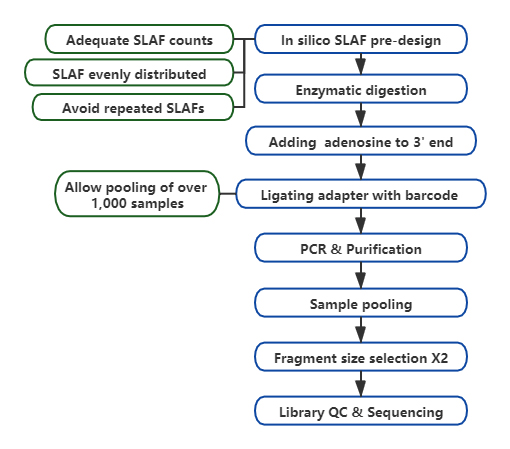
Technesch Workflow
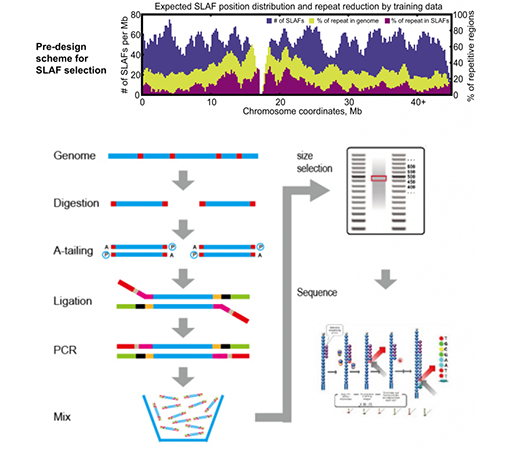
SLAF vs Bestehend RRL Methoden
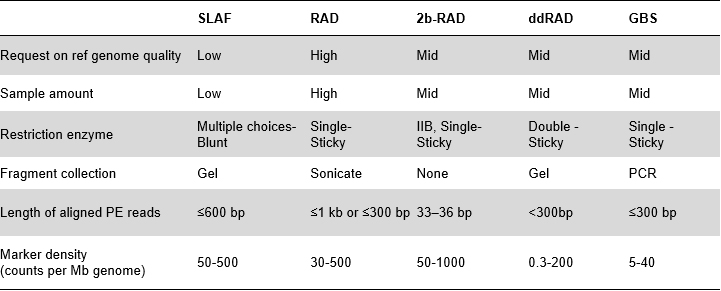
Virdeeler vun SLAF
Méi héich genetesch Marker Entdeckungseffizienz- Kombinéiert mat High-Throughput Sequencing Technologie, konnt SLAF-Seq Honnerte vun Dausende vun Tags erreechen, déi am ganze Genom entdeckt goufen, fir d'Ufro vu verschiddenen Fuerschungsprojeten ze erfëllen, entweder mat oder ouni Referenzgenom.
Benotzerdefinéiert & flexibel experimentell Design- Fir verschidde Fuerschungsziler oder Aarte sinn verschidden enzymatesch Verdauungsstrategien verfügbar, dorënner Single-Enzym, Dual-Enzyme a Multi-Enzym Verdauung.Verdauungsstrategie gëtt am Silico virbewäert fir en optimalen Enzymdesign ze garantéieren.
Héich Effizienz an enzymatesche Verdauung- Pre-entworf enzymatesch Verdauung liwwert méi gläichméisseg verdeelt SLAFs op Chromosomen.Fragmentsammlung effizient kann iwwer 95% erreechen.
Vermeiden repetitive Sequenz- Prozentsaz vun repetitive Sequenz an SLAF-Seq Daten gëtt op manner wéi 5% reduzéiert, besonnesch an Arten mat héijen Niveau vun repetitive Elementer, wéi Weess, Mais, etc.
Selbst entwéckelt bioinformatesche Workflow- BMK huet en integréierte bioinformatesche Workflow entwéckelt fir SLAF-Seq Technologie fir Zouverlässegkeet an Genauegkeet vum finalen Output ze garantéieren.
Uwendung vun SLAF
Genetesch Verbindungskaart
Héich-Dicht genetesch Kaart Konstruktioun an Identifikatioun vu Loci, déi Blummearteigenschaften am Chrysanthemum kontrolléieren (Chrysanthemum x morifolium Ramat.)
Journal: Horticulture Research Verëffentlecht: 2020.7

GWAS
Identifikatioun vun engem Kandidat Gen assoziéiert mam Isofavone Inhalt a Sojabohnen Somen mat Genom-breet Associatioun a Verknüpfungsmapping
Journal: The Plant Journal Verëffentlecht: 2020.08

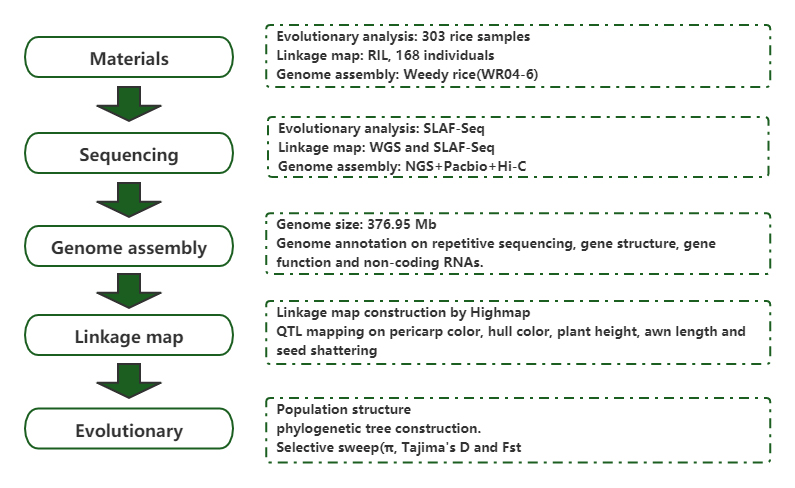
Evolutionär Genetik
Bevëlkerungsgenomesch Analyse an de novo Assemblée verroden den Urspronk vum weedy Reis als evolutivt Spill
Journal: Molecular Plant Verëffentlecht: 2019.5
Bulked Segregant Analyse (BSA)
GmST1, déi eng Sulfotransferase codéiert, gëtt Resistenz géint Soja Mosaik Virus Stämme G2 a G3
Journal: Plant, Cell & Environment Verëffentlecht: 2021.04

Referenz
Sun X, Liu D, Zhang X, et al.SLAF-Seq: eng effizient Method vu grousser Skala de novo SNP Entdeckung a Genotyping mat High-Throughput Sequencing [J].Plos eent, 2013, 8 (3): e58700
Song X, Xu Y, Gao K, et al.Héich-Dicht genetesch Kaart Konstruktioun an Identifikatioun vun Loci Kontroll Blumme-Typ Spure am Chrysanthemum (Chrysanthemum × morifolium Ramat.).Hortic Res.2020;7:108.
Wu D, Li D, Zhao X, et al.Identifikatioun vun engem Kandidat Gen assoziéiert mam Isoflavon Inhalt an Sojabohnen Somen mat Genome-breet Associatioun a Verknëppungsmapping.Planz J. 2020;104 (4): 950-963.
Sun J, Ma D, Tang L, et al.Populatiounsgenomesch Analyse an De Novo Assemblée verroden den Urspronk vum Weedy Rice als evolutivt Spill.Mol Plant.2019;12(5):632-647.Mol Plant.2018;11(11):1360-1376.
Zhao X, Jing Y, Luo Z, et al.GmST1, déi eng Sulfotransferase codéiert, gëtt Resistenz géint Soja Mosaik Virus Stämme G2 a G3.Planz Zell Ëmfeld.2021; 10.1111 / Stéck. 14066
Post Zäit: Jan-04-2022