

特定遺伝子座増幅断片シーケンス (SLAF-Seq)





サービス内容
技術スキーム
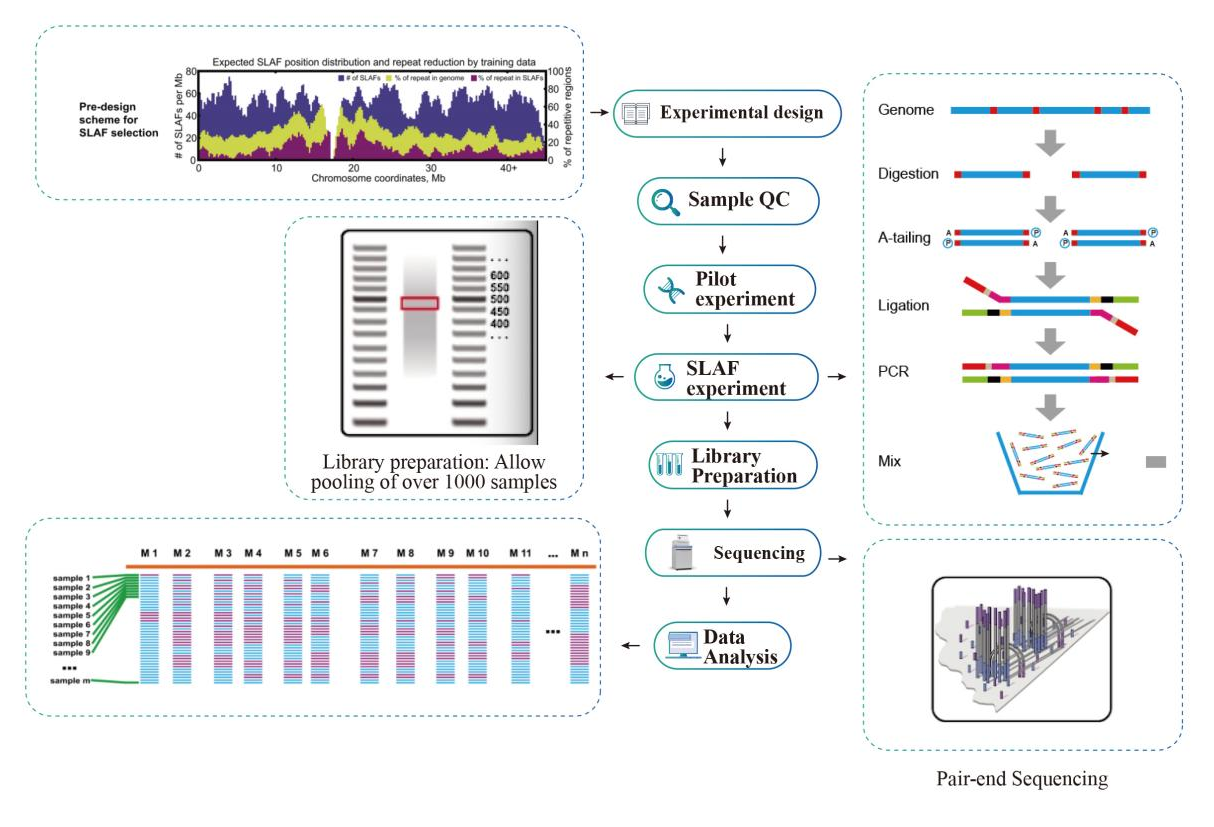
作業の流れ

サービスのメリット
高いマーカー発見効率- ハイスループットシーケンシング技術は、SLAF-Seq がゲノム全体内の数十万のタグを発見するのを支援します。
ゲノムへの依存性が低い- 参照ゲノムの有無にかかわらず、種に適用できます。
柔軟なスキーム設計- 単一酵素、二重酵素、複数酵素による消化、およびさまざまな種類の酵素をすべて選択して、さまざまな研究目標や種に対応できます。最適な酵素設計を保証するために、コンピュータでの事前評価が使用されます。
効率的な酵素消化- 条件を最適化するために事前実験が実行され、正式な実験が安定して信頼できるものになります。フラグメント収集効率は 95% 以上を達成できます。
均等に分散された SLAF タグ- SLAF タグはすべての染色体に最大限に均等に分布しており、平均 4 kb あたり 1 つの SLAF を実現します。
繰り返しを効果的に回避する- SLAF-Seq データの反復配列は、特に小麦、トウモロコシなどの反復レベルが高い種で 5% 未満に減少します。
豊富な経験-植物、哺乳類、鳥、昆虫、水生生物などを含む数百の種に関する2000以上のクローズされたSLAF-Seqプロジェクト。
自社開発のバイオインフォマティクスワークフロー- 最終出力の信頼性と精度を確保するために、SLAF-Seq 用の統合バイオインフォマティクス ワークフローが BMKGENE によって開発されました。
サービス仕様
| プラットホーム | 濃度(ng/gl) | 合計 (ug) | OD260/280 |
| イルミナ ノヴァシーク | >35 | >1.6(ボリューム>15μl) | 1.6~2.5 |
推奨されるシーケンス戦略
シーケンス深度: 10X/タグ
| ゲノムサイズ | 推奨される SLAF タグ |
| < 500MB | 100K または WGS |
| 500MB~1GB | 100K |
| 1GB~2GB | 200K |
| 巨大または複雑なゲノム | 300 - 400K |
| アプリケーション
| 推奨 人口規模
| シーケンス戦略と深さ
| |
| 深さ
| タグ番号
| ||
| GWAS
| サンプル数 ≥ 200
| 10倍
|
によると ゲノムサイズ
|
| 遺伝子進化
| それぞれの個体 サブグループ ≥ 10; 合計サンプル数 ≥ 30
| 10倍
| |
推奨されるサンプル配信
容器:2ml遠沈管
ほとんどのサンプルは、エタノール中で保存しないことをお勧めします。
サンプルのラベル付け: サンプルには明確にラベルが付けられ、提出されたサンプル情報フォームと同一である必要があります。
発送: ドライアイス: サンプルは最初にバッグに梱包し、ドライアイスに埋める必要があります。
サービスのワークフロー






サンプルQC
パイロット実験
SLAF実験
ライブラリの準備
シーケンス
データ分析
アフターサービス
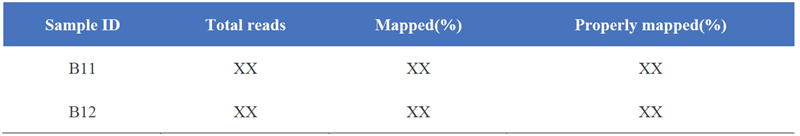
1. マップ結果の統計

2. SLAFマーカーの開発

3. バリエーションの注釈

| 年 | ジャーナル | IF | タイトル | アプリケーション |
| 2022年 | 自然コミュニケーション | 17.694 | 牡丹のギガ染色体とギガゲノムのゲノム基盤 シャクヤク | SLAF-GWAS |
| 2015年 | 新しい植物学者 | 7.433 | 家畜化の足跡は、農学的に重要なゲノム領域を固定している 大豆 | SLAF-GWAS |
| 2022年 | 先端研究ジャーナル | 12.822 | Gossypium barbadense の G. hirsutum へのゲノムワイド人工遺伝子移入 綿繊維の品質と収量を同時に改善するための優れた遺伝子座を明らかにする 特徴 | SLAF-進化遺伝学 |
| 2019年 | 分子植物 | 10.81 | 集団ゲノム解析とDe NovoアセンブリでWeedyの起源が明らかに 進化ゲームとしての米 | SLAF-進化遺伝学 |
| 2019年 | 自然遺伝学 | 31.616 | コイ、Cyprinus cario のゲノム配列と遺伝的多様性 | SLAF-リンケージマップ |
| 2014年 | 自然遺伝学 | 25.455 | 栽培された落花生のゲノムは、マメ科植物の核型、倍数体に関する洞察を提供します。 進化と作物の栽培化。 | SLAF-リンケージマップ |
| 2022年 | 植物バイオテクノロジージャーナル | 9.803 | ST1の同定により、種子の形態のヒッチハイクを伴う選択が明らかになった 大豆栽培時の油分と | SLAF-Markerの開発 |
| 2022年 | 国際分子科学ジャーナル | 6.208 | コムギ Leymus mollis 2Ns の同定と DNA マーカー開発 (2D) 二染色体染色体置換 | SLAF-Markerの開発 |





