

Sequenziamento del genoma de novo di piante/animali




Vantaggi del servizio
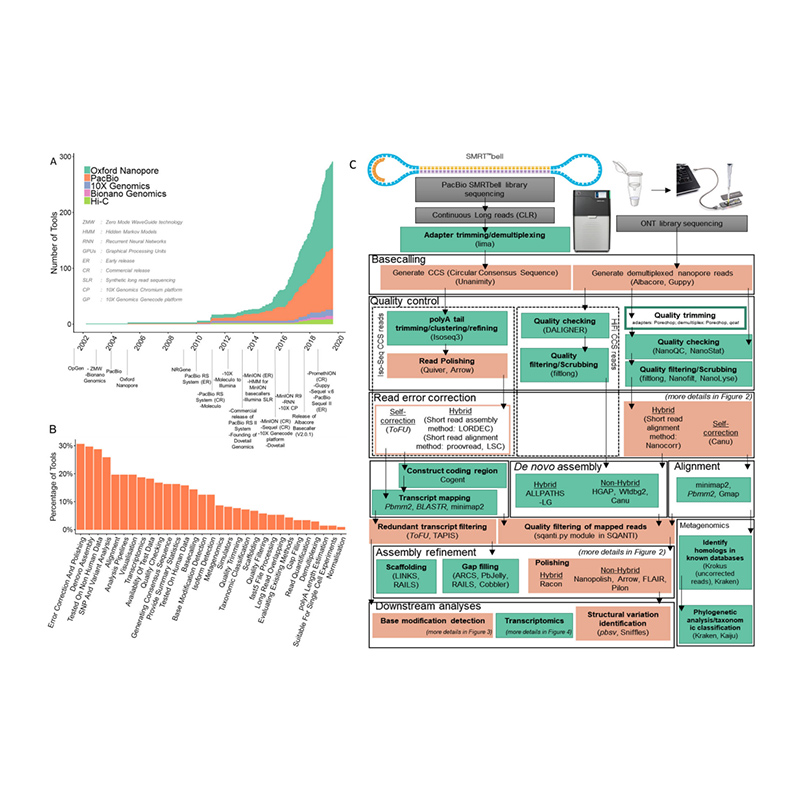
Sviluppo di piattaforme di sequenziamento e bioinformatica inde novoassemblaggio del genoma
(Amarasinghe SL et al.,Biologia del genoma, 2020)
● Costruire nuovi genomi e migliorare i genomi di riferimento esistenti per le specie di interesse.
● Maggiore precisione, continuità e completezza nell'assemblaggio
● Costruire risorse fondamentali per la ricerca sul polimorfismo delle sequenze, sui QTL, sull'editing genetico, sull'allevamento, ecc.
● Dotato di una gamma completa di piattaforme di sequenziamento di terza generazione: soluzione completa per l'assemblaggio del genoma
● Strategie flessibili di sequenziamento e assemblaggio che soddisfano diversi genomi con caratteristiche diverse
● Team di bioinformatici altamente qualificati con grande esperienza in complessi assemblaggi di genomi, inclusi poliploidi, genomi giganti, ecc.
● Oltre 100 casi di successo con un fattore di impatto pubblicato cumulativo di oltre 900
● Tempi di consegna di soli 3 mesi per l'assemblaggio del genoma a livello cromosomico.
● Solido supporto tecnico con una serie di brevetti e copyright di software sia in ambito sperimentale che bioinformatico.
Specifiche del servizio
|
Contenuto
|
piattaforma
|
Leggi Lunghezza
|
Copertura
|
| Indagine sul genoma
| Illumina NovaSeq
| PE150
| ≥ 50X
|
| Sequenziamento del genoma
| PacBio Revio
| Letture HiFi da 15 kb
| ≥ 30X
|
| Ciao-C
| Illumina NovaSeq
| PE150
| ≥100X
|
Flusso di lavoro

Requisiti e consegna del campione
Requisiti del campione:
| Specie | Tessuto | Per PacBio | Per Nanoporo |
| Animali | Organi viscerali (fegato, milza, ecc.) | ≥ 1,0 g | ≥ 3,5 g |
| Muscolo | ≥ 1,5 g | ≥ 5,0 g | |
| Sangue di mammiferi | ≥ 1,5ml | ≥ 5,0ml | |
| Sangue di pesci o uccelli | ≥ 0,2ml | ≥ 0,5ml | |
| Impianti | Foglie fresche | ≥ 1,5 g | ≥ 5,0 g |
| Petalo o stelo | ≥ 3,5 g | ≥ 10,0 g | |
| Radici o semi | ≥ 7,0 g | ≥ 20,0 g | |
| Celle | Coltura cellulare | ≥ 3×107 | ≥1×108 |
Consegna del campione consigliata
Contenitore: provetta da centrifuga da 2 ml (la carta stagnola non è consigliata)
Per la maggior parte dei campioni si consiglia di non conservare in etanolo.
Etichettatura dei campioni: i campioni devono essere chiaramente etichettati e identici al modulo informativo del campione inviato.
Spedizione: Ghiaccio secco: i campioni devono essere prima imballati in sacchetti e sepolti nel ghiaccio secco.
Flusso di lavoro del servizio

Progettazione dell'esperimento

Consegna del campione

Estrazione del DNA

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita
*I risultati dimostrativi mostrati qui provengono tutti da genomi pubblicati con Biomarker Technologies
1.Circos sull'assemblaggio del genoma a livello cromosomico diG. rotundifoliodalla piattaforma di sequenziamento Nanopore
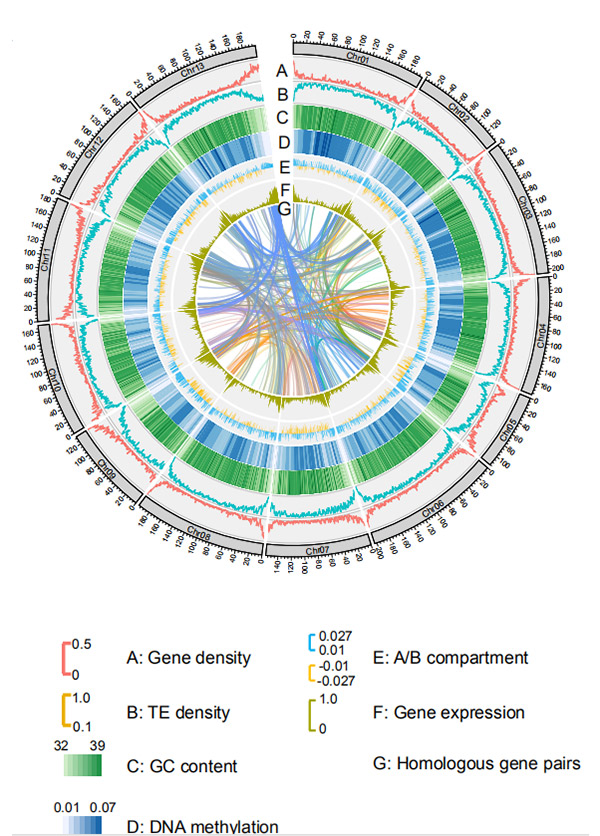
Wang M et al.,Biologia Molecolare ed Evoluzione, 2021
2. Statistiche sull'assemblaggio e annotazione del genoma della segale Weining
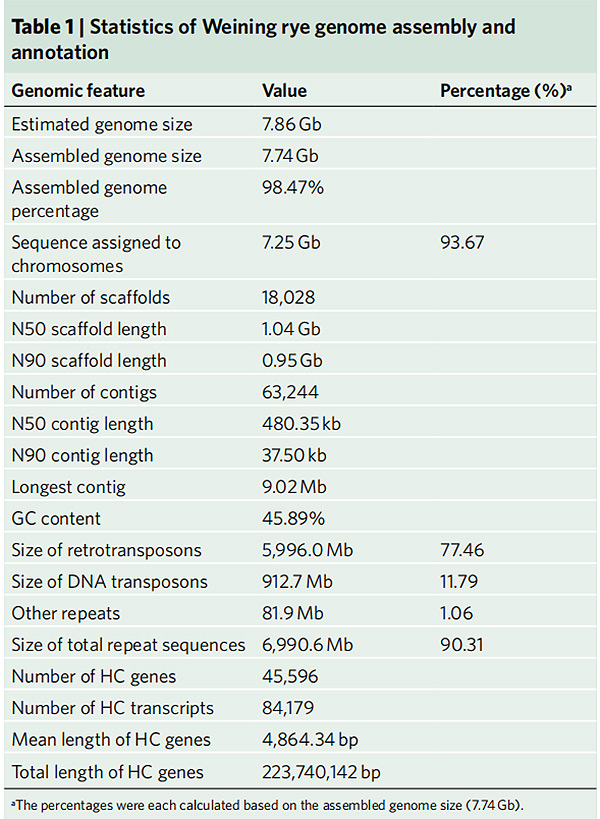
Li G et al.,Genetica della natura, 2021
3.Previsione genetica diSechium edulegenoma, derivato da tre metodi di previsione:Di nuovoprevisione, previsione basata sull'omologia e previsione basata sui dati RNA-Seq
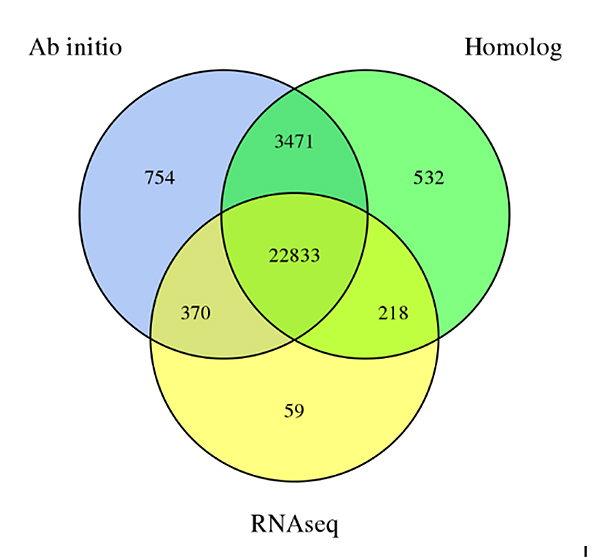
Fu A et al.,Ricerca sull'orticoltura, 2021
4.Identificazione di ripetizioni terminali lunghe intatte in tre genomi di cotone
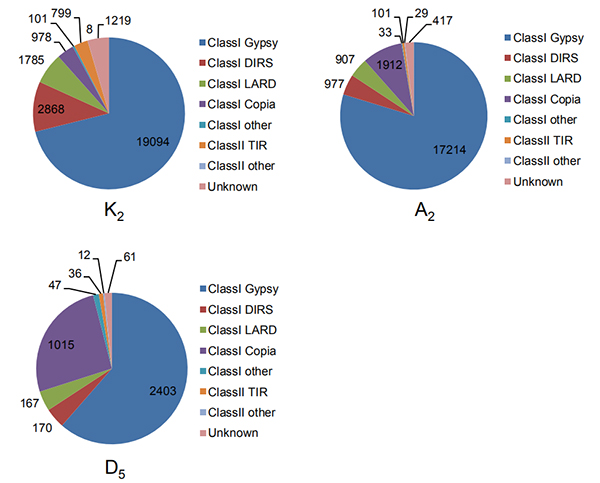
Wang M et al.,Biologia Molecolare ed Evoluzione, 2021
5.Mappa termica Hi-C delC. acuminatagenoma che mostra interazioni generali a livello dell'intero genoma.L'intensità delle interazioni Hi-C è proporzionale alla distanza lineare tra i contigui.Una linea retta pulita su questa mappa termica indica un ancoraggio estremamente accurato dei contigu sui cromosomi.(Rapporto di ancoraggio Contig: 96,03%)
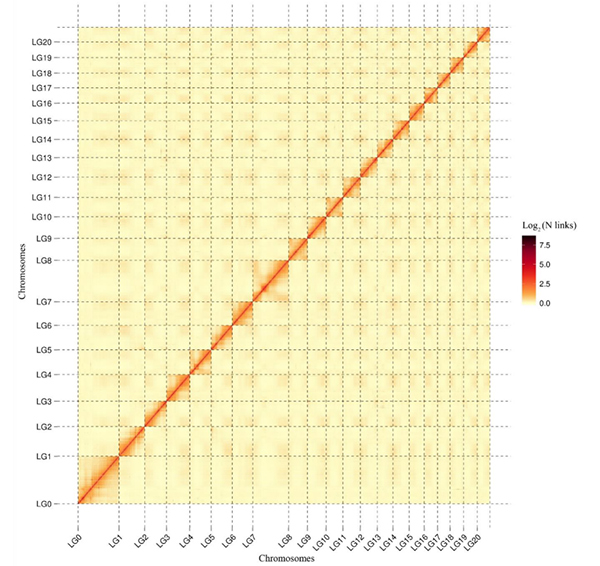
Kang M et al.,Comunicazioni sulla natura,2021
Caso BMK
Un assemblaggio del genoma di alta qualità evidenzia le caratteristiche genomiche della segale e i geni agronomicamente importanti
Pubblicato: Genetica della natura, 2021
Strategia di sequenziamento:
Assemblaggio del genoma: modalità PacBio CLR con libreria da 20 kb (497 Gb, circa 63×)
Correzione della sequenza: NGS con libreria di DNA da 270 bp (430 Gb, circa 54×) su piattaforma Illumina
Ancoraggio Contigs: libreria Hi-C (560 Gb, circa 71×) su piattaforma Illumina
Carta ottica: (779,55 Gb, ca. 99×) su Bionano Irys
Risultati chiave
1. È stato pubblicato un insieme del genoma della segale Weining con una dimensione totale del genoma di 7,74 Gb (98,74% della dimensione del genoma stimata mediante citometria a flusso).L'impalcatura N50 di questo assieme ha raggiunto 1,04 Gb.Il 93,67% dei contig era ancorato con successo su 7 pseudo-cromosomi.Questo insieme è stato valutato mediante la mappa di collegamento, LAI e BUSCO, che ha prodotto punteggi elevati in tutte le valutazioni.
2.Ulteriori studi sulla genomica comparativa, sulla mappa del collegamento genetico e sugli studi di trascrittomica sono stati condotti sulla base di questo genoma.Sono state rivelate una serie di caratteristiche genomiche correlate ai tratti, tra cui duplicazioni geniche a livello dell'intero genoma e il loro impatto sui geni della biosintesi dell'amido;organizzazione fisica dei complessi loci della prolamina, caratteristiche dell'espressione genica alla base del tratto iniziale della voce e presunte regioni cromosomiche e loci associati all'addomesticamento nella segale.
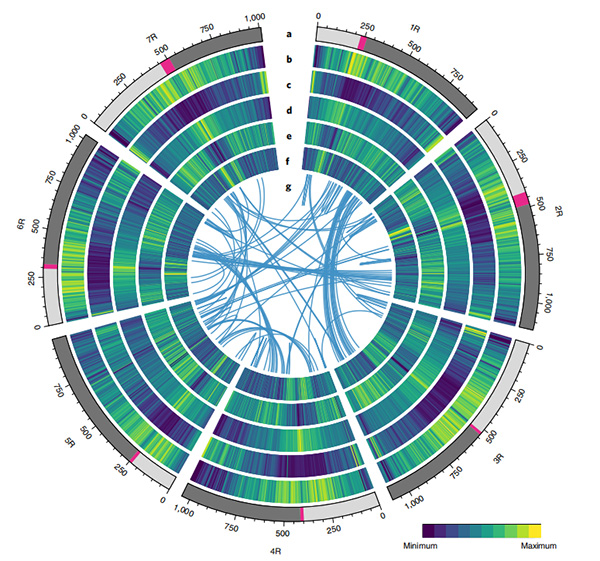 Diagramma Circos sulle caratteristiche genomiche del genoma della segale Weining | 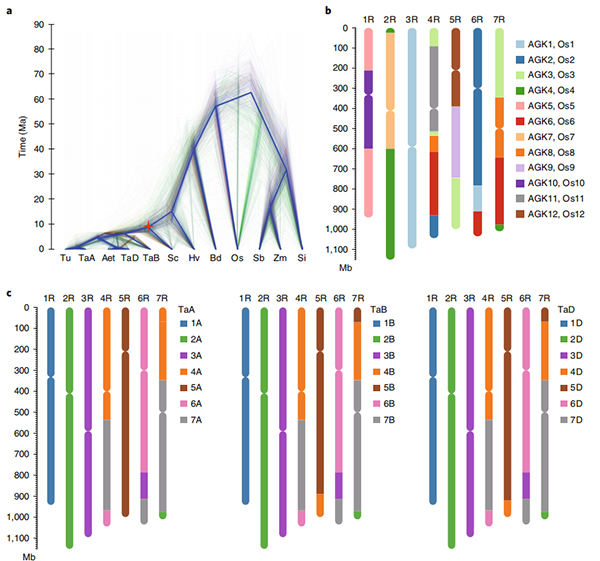 Analisi di sintesi evolutiva e cromosomica del genoma della segale |
Li, G., Wang, L., Yang, J.et al.Un assemblaggio del genoma di alta qualità evidenzia le caratteristiche genomiche della segale e i geni agronomicamente importanti.Nat Genet 53,574–584 (2021).
https://doi.org/10.1038/s41588-021-00808-z





