

Assemblaggio del genoma basato su Hi-C







Vantaggi del servizio
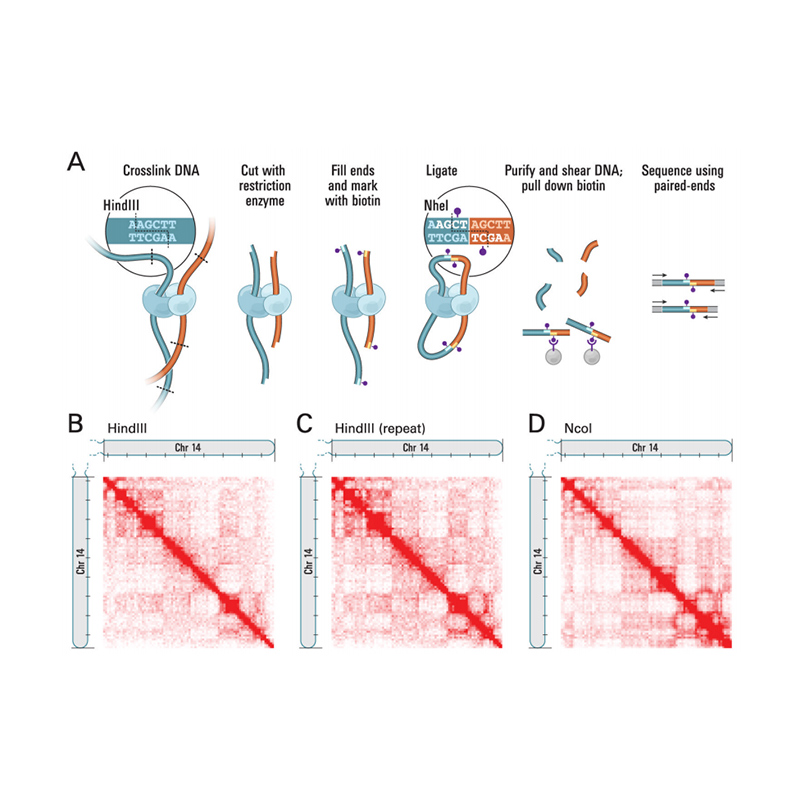
Panoramica di Hi-C
(Lieberman-Aiden E et al.,Scienza, 2009)
● Non è necessario costruire una popolazione genetica per l'ancoraggio dei contig;
● Una maggiore densità di marcatori porta a un rapporto di ancoraggio dei contig più elevato, superiore al 90%;
● Consente la valutazione e la correzione degli insiemi di genomi esistenti;
● Tempi di consegna più brevi con maggiore precisione nell'assemblaggio del genoma;
● Esperienza ricca con oltre 1000 librerie Hi-C costruite per oltre 500 specie;
● Oltre 100 casi di successo con un Impact Factor cumulativo pubblicato superiore a 760;
● Assemblaggio del genoma basato su Hi-C per genoma poliploide, tasso di ancoraggio del 100% raggiunto nel progetto precedente;
● Brevetti interni e copyright di software per esperimenti Hi-C e analisi dei dati;
● Software di ottimizzazione dei dati visualizzati sviluppato internamente, consente lo spostamento, l'inversione, la revoca e la ripetizione manuale dei blocchi.
Specifiche del servizio
|
Tipo di libreria
|
piattaforma | Leggi Lunghezza | Consiglia strategia |
| Ciao-C | Illumina NovaSeq | PE150 | ≥ 100X |
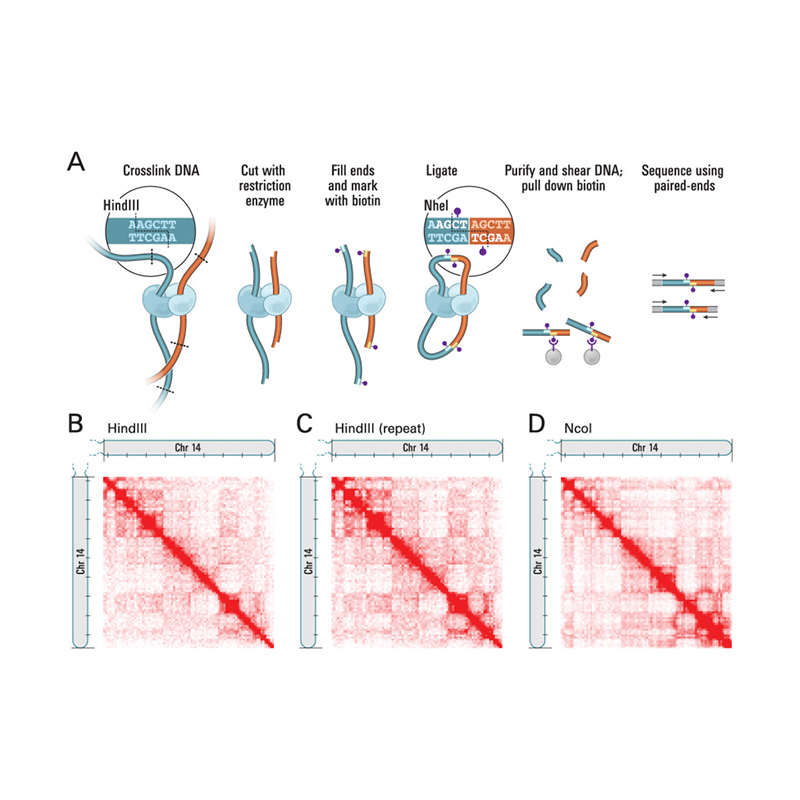
Analisi bioinformatiche
● Controllo della qualità dei dati grezzi
● Controllo di qualità della libreria Hi-C
● Assemblaggio del genoma basato su Hi-C
● Valutazione post-assemblaggio
Requisiti e consegna del campione
Requisiti del campione:
| Animale | Fungo | Impianti
|
| Tessuto congelato: 1-2 g per libreria Celle: 1x 10^7 celle per libreria | Tessuto congelato: 1 g per libreria | Tessuto congelato: 1-2 g per libreria
|
| *Si consiglia vivamente di inviare almeno 2 aliquote (1 g ciascuna) per l'esperimento Hi-C. | ||
Consegna del campione consigliata
Contenitore: provetta da centrifuga da 2 ml (la carta stagnola non è consigliata)
Per la maggior parte dei campioni si consiglia di non conservare in etanolo.
Etichettatura dei campioni: i campioni devono essere chiaramente etichettati e identici al modulo informativo del campione inviato.
Spedizione: Ghiaccio secco: i campioni devono essere prima imballati in sacchetti e sepolti nel ghiaccio secco.
Flusso di lavoro del servizio

Progettazione dell'esperimento

Consegna del campione

Estrazione del DNA

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita
*I risultati dimostrativi mostrati qui provengono tutti da genomi pubblicati con Biomarker Technologies
1.Mappa termica di interazione Hi-C diCamptotheca acuminatagenoma.Come mostrato sulla mappa, l'intensità delle interazioni è negativamente correlata alla distanza lineare, che indica un assemblaggio a livello cromosomico altamente accurato.(Rapporto di ancoraggio: 96,03%)
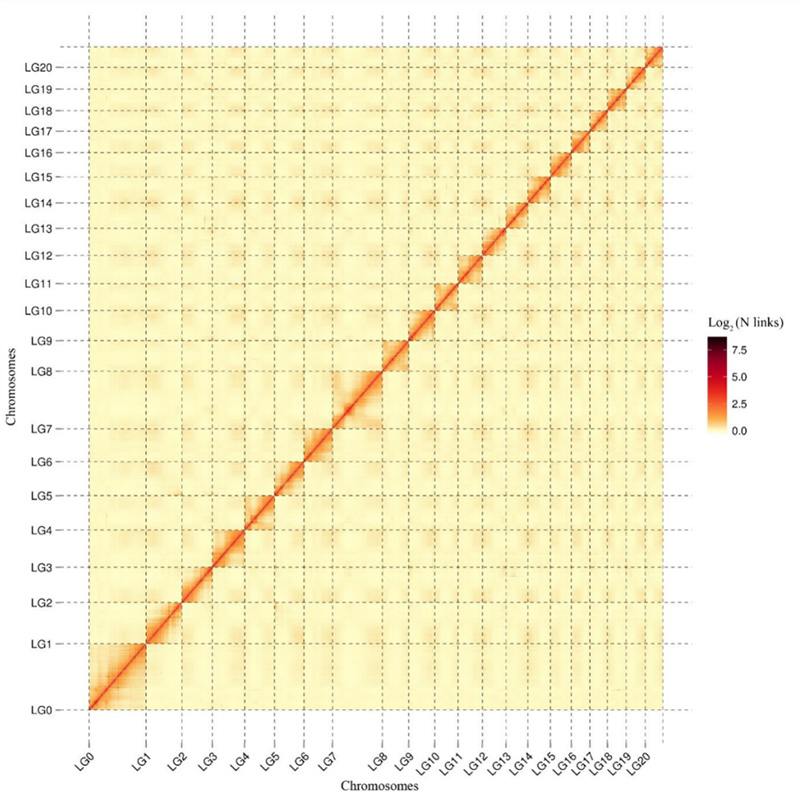
Kang M et al.,Comunicazioni sulla natura, 2021
2.Hi-C ha facilitato la convalida delle inversioni traGossypium hirsutumL.TM-1 A06 eG. arboreumChr06
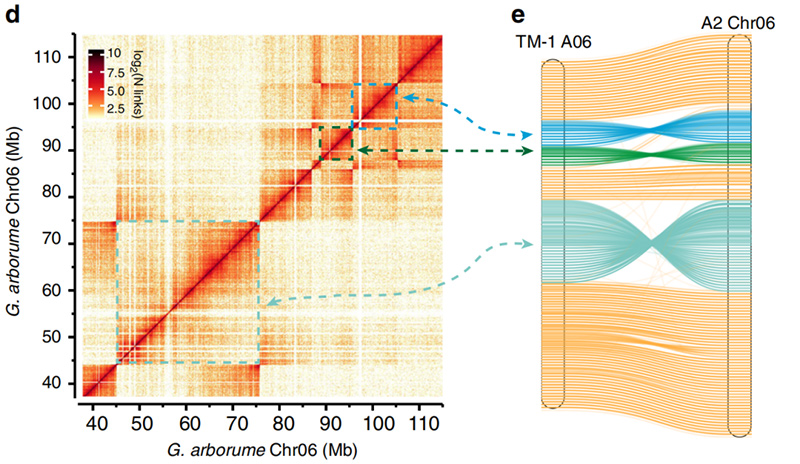
Yang Z et al.,Comunicazioni sulla natura, 2019
3.Assemblaggio e differenziazione biallelica del genoma della manioca SC205.La mappa termica Hi-C mostrava una chiara divisione nei cromosomi omologhi.
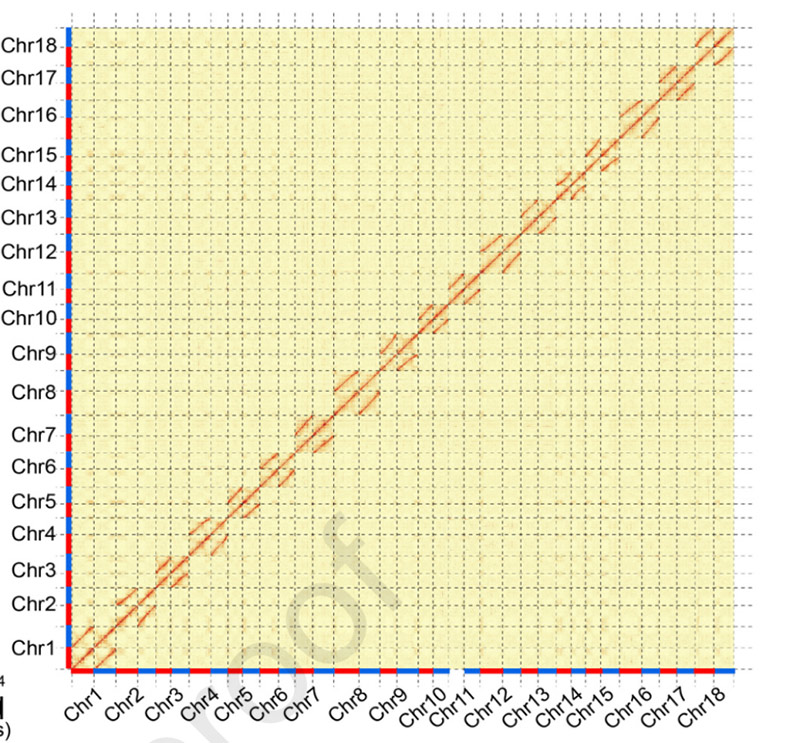
Hu W et al.,Pianta molecolare, 2021
4. Mappa termica Hi-C sull'assemblaggio del genoma di due specie di Ficus:F.microcarpa(rapporto di ancoraggio: 99,3%) eF.hispida (rapporto di ancoraggio: 99,7%)
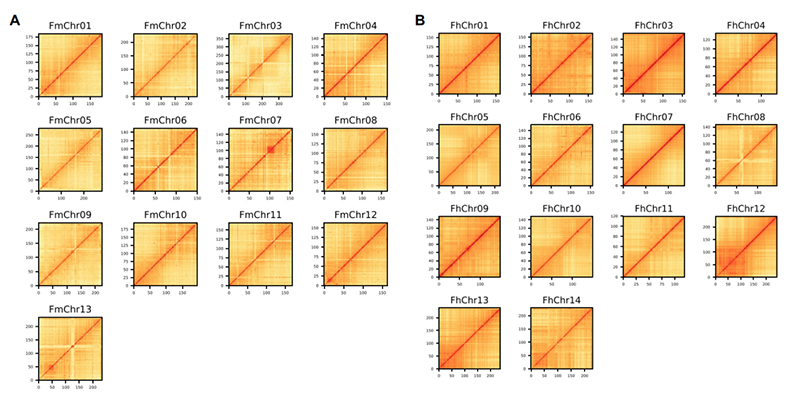
Zhang X et al.,Cellula, 2020
Caso BMK
I genomi dell'albero di banyan e della vespa impollinatrice forniscono informazioni sulla coevoluzione della vespa del fico
Pubblicato: Cellula, 2020
Strategia di sequenziamento:
F. microcarpa genoma: ca.84 X PacBio RSII (36,87 Gb) + Hi-C (44 Gb)
F. hispidagenoma: ca.97 X PacBio RSII (36,12 Gb) + Hi-C (60 Gb)
Eupristina verticillatagenoma: ca.170 PacBio RSII (65 Gb)
Risultati chiave
1.Due genomi di alberi di banyan e un genoma di vespa impollinatrice sono stati costruiti utilizzando il sequenziamento PacBio, Hi-C e la mappa di collegamento.
(1)F. microcarpagenoma: è stato stabilito un insieme di 426 Mb (97,7% della dimensione stimata del genoma) con contig N50 di 908 Kb, punteggio BUSCO del 95,6%.In totale, 423 sequenze Mb sono state ancorate a 13 cromosomi mediante Hi-C.L'annotazione del genoma ha prodotto 29.416 geni codificanti proteine.
(2)F. Hispidagenoma: è stato prodotto un insieme di 360 Mb (97,3% della dimensione stimata del genoma) con contig N50 di 492 Kb e punteggio BUSCO del 97,4%.Un totale di 359 sequenze Mb sono state ancorate su 14 cromosomi mediante Hi-C e altamente identiche alla mappa di collegamento ad alta densità.
(3)Eupristina verticillatagenoma: è stato stabilito un insieme di 387 Mb (dimensione stimata del genoma: 382 Mb) con contig N50 di 3,1 Mb e punteggio BUSCO del 97,7%.
2. L'analisi genomica comparativa ha rivelato un gran numero di variazioni strutturali tra i dueFicusgenomi, che hanno fornito risorse genetiche inestimabili per gli studi sull’evoluzione adattiva.Questo studio, per la prima volta, ha fornito approfondimenti sulla coevoluzione della vespa Fig a livello genomico.
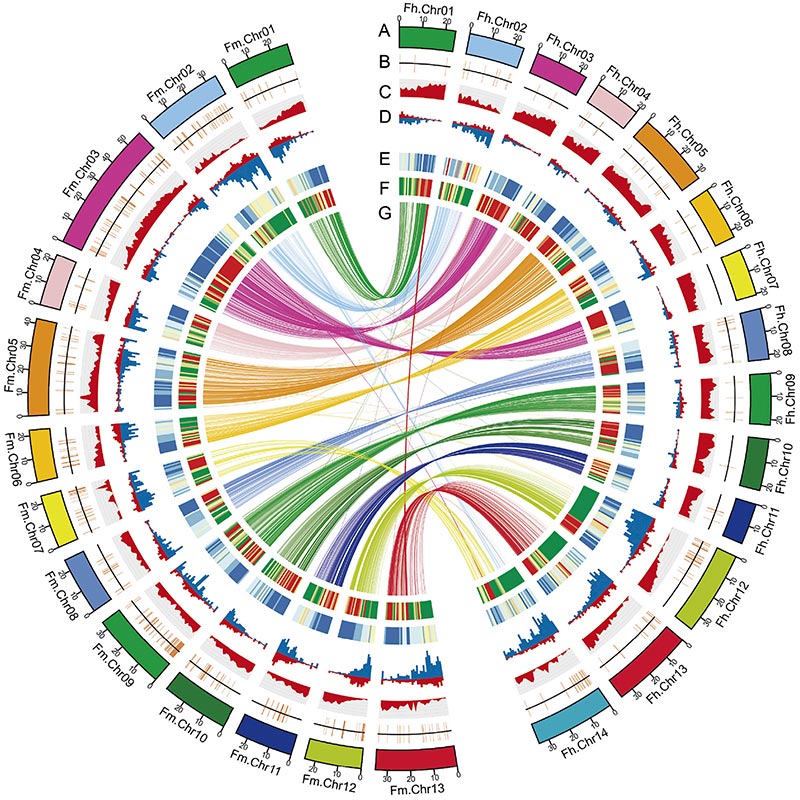 Diagramma di Circo sulle caratteristiche genomiche di dueFicusgenomi, inclusi cromosomi, duplicazioni segmentali (SD), trasposoni (LTR, TE, DNA TE), espressione genica e sintesi | 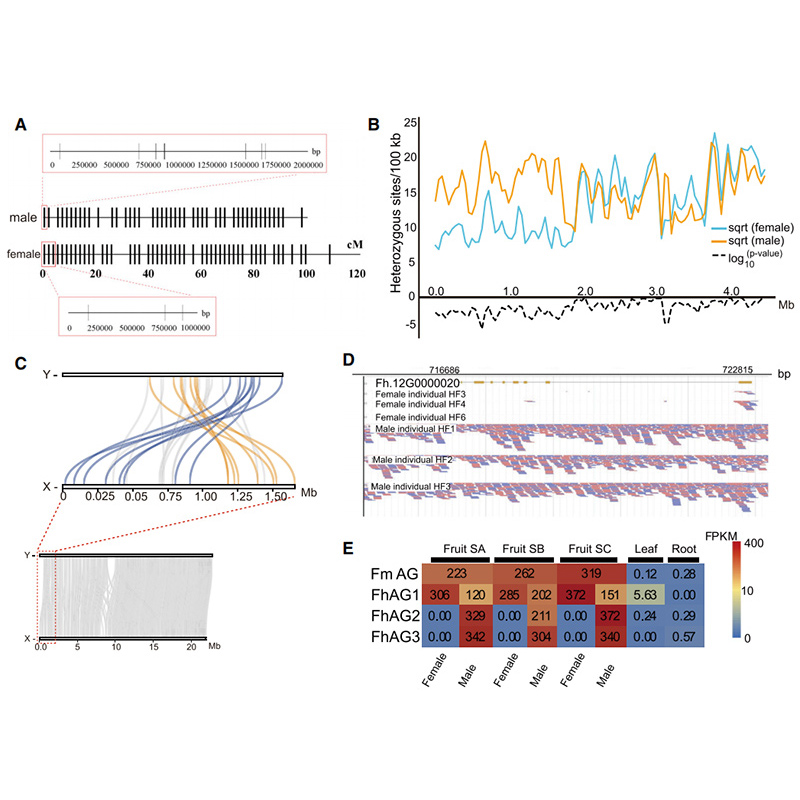 Identificazione del cromosoma Y e gene candidato alla determinazione del sesso |
Zhang, X., et al."I genomi dell'albero di banyan e della vespa impollinatrice forniscono informazioni sulla coevoluzione della vespa fico".Cella 183.4(2020).





