

Genetica evolutiva








Vantaggi del servizio
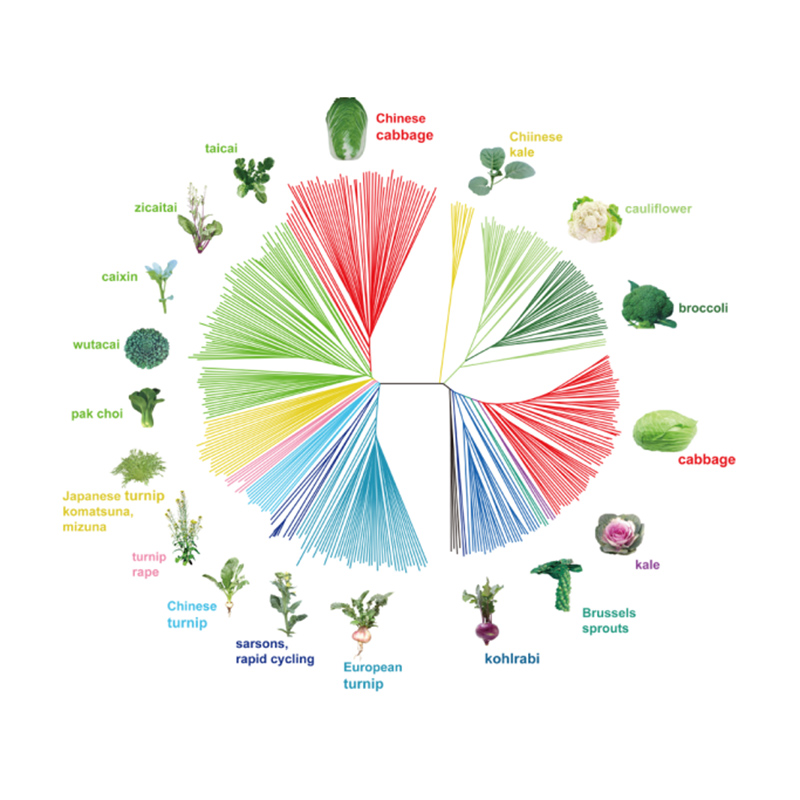
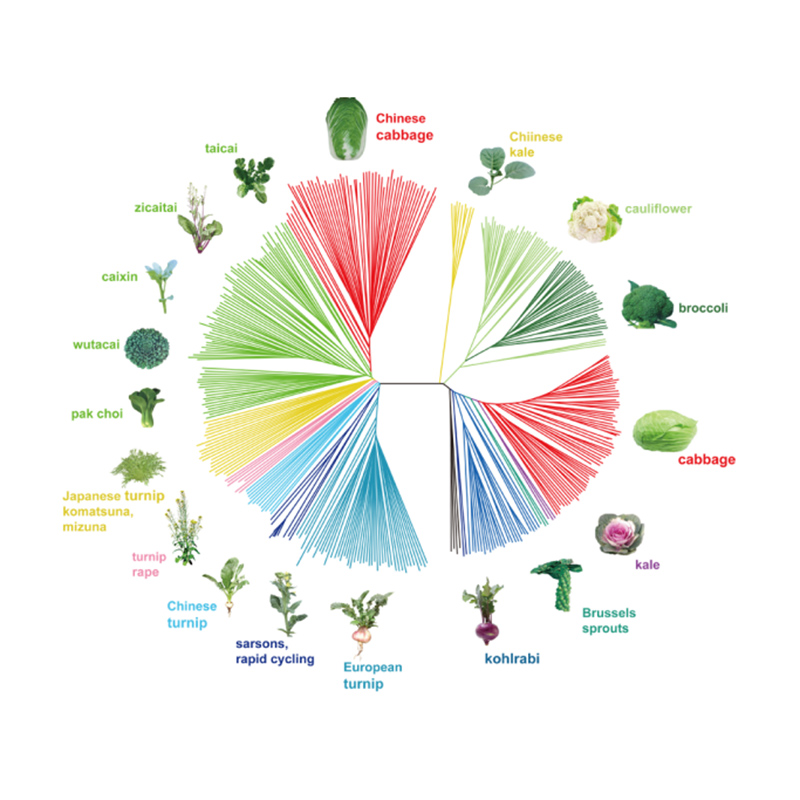
Takagi et al.,Il diario delle piante, 2013
● Stima del tempo e della velocità di divergenza delle specie in base alle variazioni a livello di nucleotidi e aminoacidi
● Rivelazione di relazioni filogenetiche più affidabili tra specie con influenza minima dell'evoluzione convergente e dell'evoluzione parallela
● Costruire collegamenti tra cambiamenti genetici e fenotipi per scoprire geni correlati ai tratti
● Stima della diversità genetica, che riflette il potenziale evolutivo delle specie
● Tempi di consegna più rapidi
● Vasta esperienza: BMK ha accumulato una vasta esperienza in progetti relativi alla popolazione e all'evoluzione per oltre 12 anni, coprendo centinaia di specie, ecc. e ha contribuito a oltre 80 progetti di alto livello pubblicati su Nature Communications, Molecular Plants, Plant Biotechnology Journal, ecc.
Specifiche del servizio
Materiali:
Normalmente si raccomandano almeno tre sottopopolazioni (ad esempio sottospecie o ceppi).Ciascuna sottopopolazione deve contenere non meno di 10 individui (Piante >15, può essere ridotto per le specie rare).
Strategia di sequenziamento:
* WGS può essere impiegato per specie con genoma di riferimento di alta qualità, mentre SLAF-Seq è applicabile a specie con o senza genoma di riferimento o genoma di riferimento di scarsa qualità.
| Applicabile alla dimensione del genoma | WGS | Tag SLAF (×10.000) |
| ≤ 500 MB | 10×/individuo | WGS è più raccomandato |
| 500 Mb - 1 Gb | 10 | |
| 1 GB - 2 GB | 20 | |
| ≥2 GB | 30 |
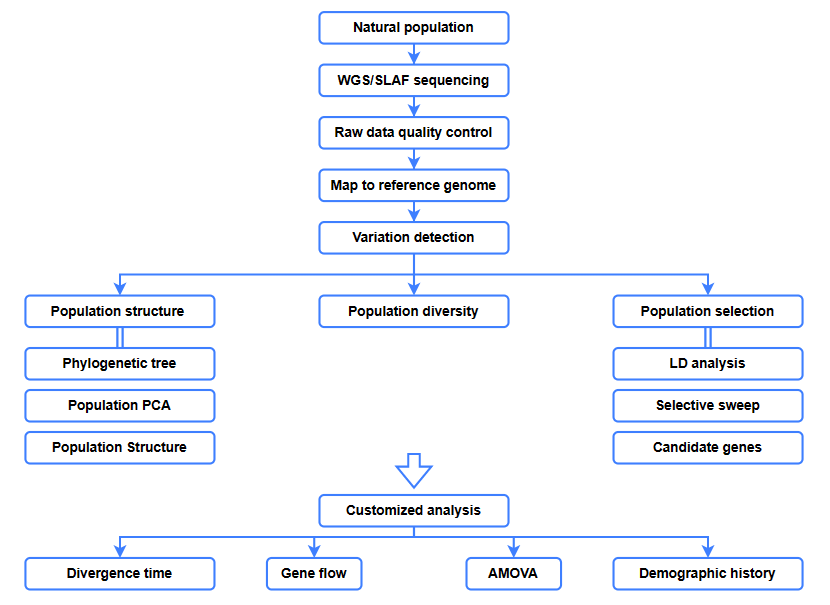
Analisi bioinformatiche
● Analisi evolutiva
● Scansione selettiva
● Flusso genico
● Storia demografica
● Tempo di divergenza
Requisiti e consegna del campione
Requisiti del campione:
| Specie | Tessuto | WGS-NGS | SLAF |
| Animale
| Tessuto viscerale |
0,5 ~ 1 g
|
0,5 g
|
| Tessuto muscolare | |||
| Sangue di mammiferi | 1,5 ml
| 1,5 ml
| |
| Sangue di pollame/pesce | |||
| Pianta
| Foglia fresca | 1~2g | 0,5 ~ 1 g |
| Petalo/stelo | |||
| Radice/seme | |||
| Celle | Cellula in coltura |
| gDNA | Concentrazione | Quantità (ug) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1.6-2.5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Flusso di lavoro del servizio

Progettazione dell'esperimento

Consegna del campione

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita
*I risultati dimostrativi mostrati qui provengono tutti da genomi pubblicati con BMKGENE
1.L'analisi dell'evoluzione contiene la costruzione dell'albero filogenetico, della struttura della popolazione e del PCA in base alle variazioni genetiche.
L'albero filogenetico rappresenta le relazioni tassonomiche ed evolutive tra specie con antenato comune.
La PCA mira a visualizzare la vicinanza tra le sottopopolazioni.
La struttura della popolazione mostra la presenza di sottopopolazioni geneticamente distinte in termini di frequenze alleliche.
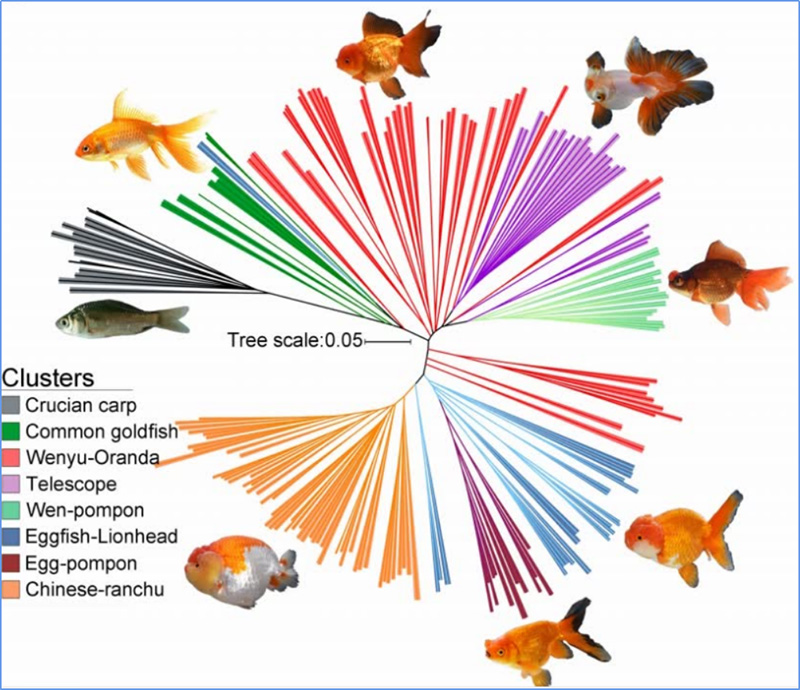
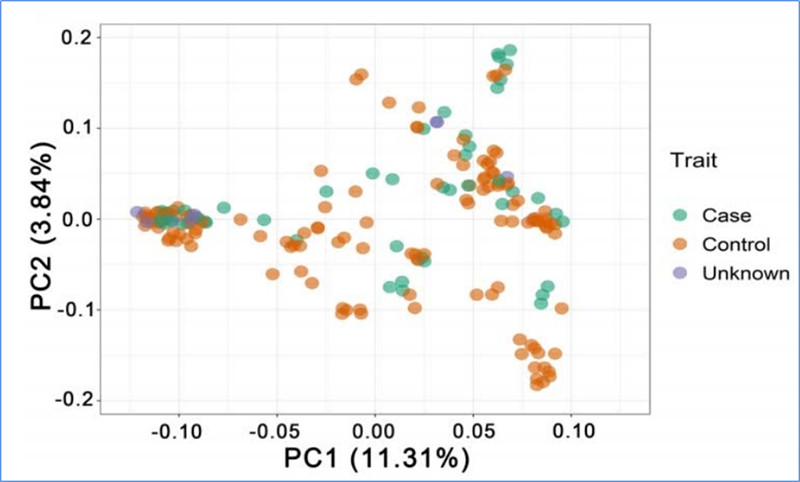
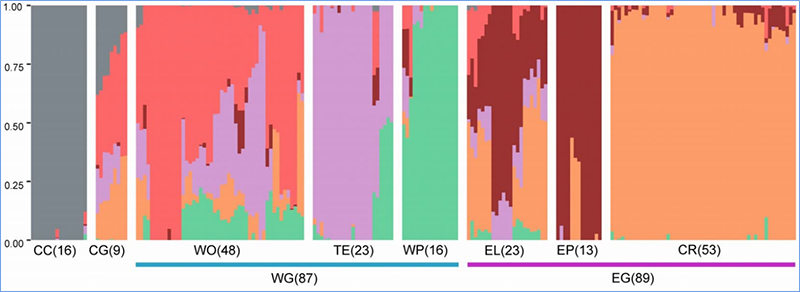
Chen, et.al.,PNAS, 2020
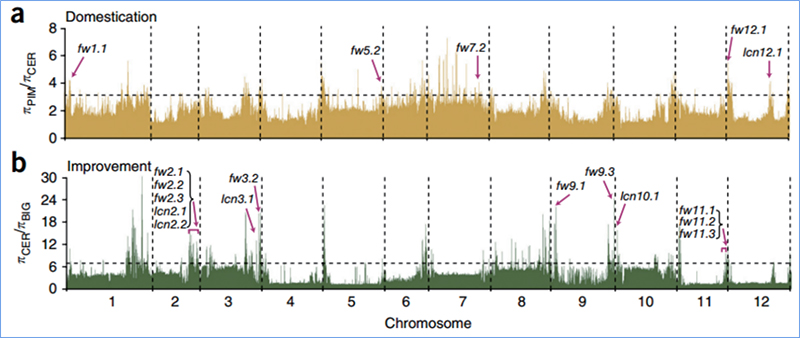
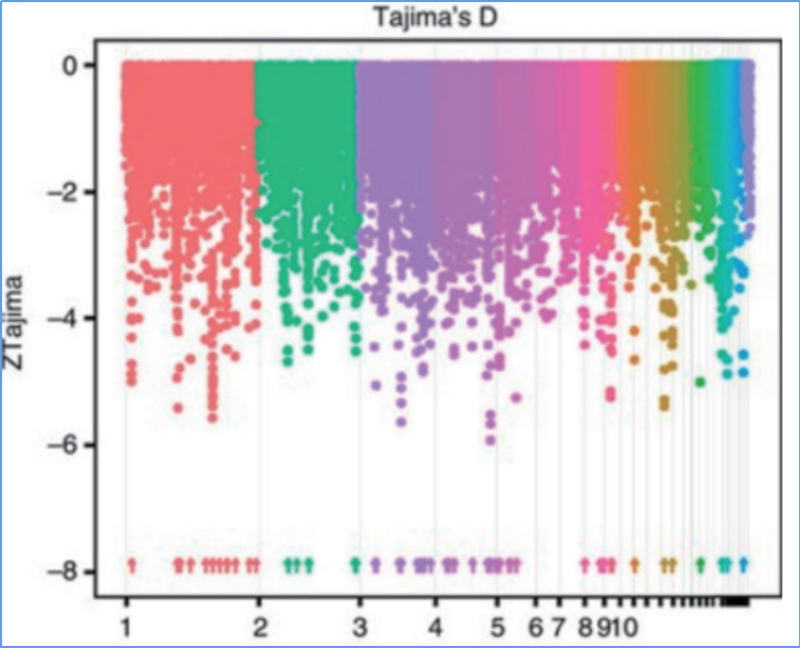
2.Scansione selettiva
La scansione selettiva si riferisce a un processo mediante il quale viene selezionato un sito vantaggioso e le frequenze dei siti neutrali collegati vengono aumentate e quelle dei siti non collegati vengono diminuite, con conseguente riduzione della regionale.
Il rilevamento dell'intero genoma su regioni di scansione selettive viene elaborato calcolando l'indice genetico della popolazione (π, Fst, D di Tajima) di tutti gli SNP all'interno di una finestra scorrevole (100 Kb) a un determinato passaggio (10 Kb).
Diversità nucleotidica (π)
D. di Tajima
Indice di fissazione (Fst)
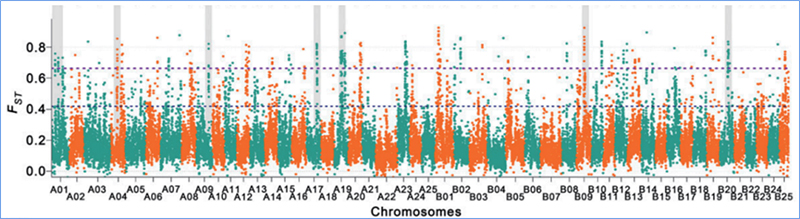
Wu, et.al.,Pianta molecolare, 2018
3.Flusso genico
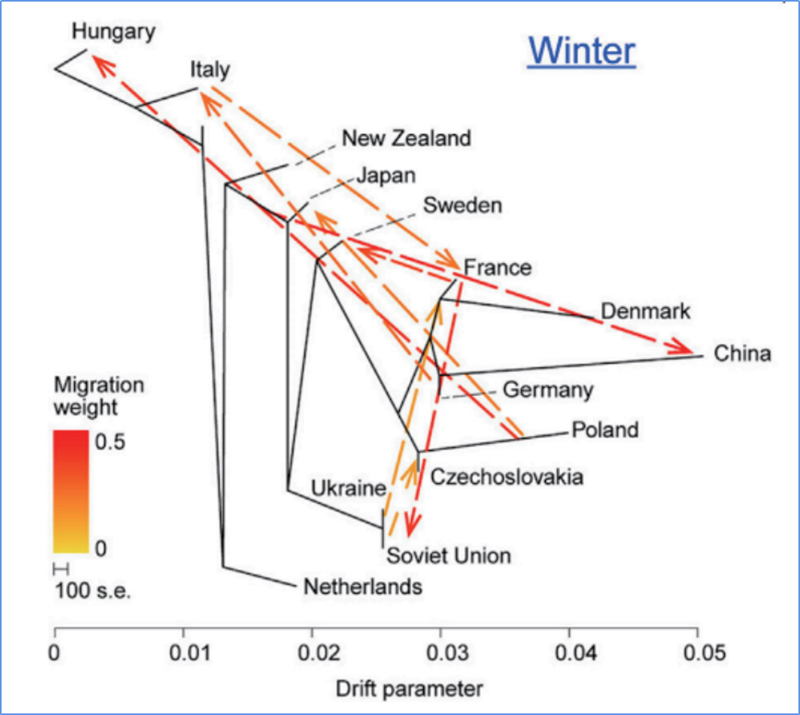
Wu, et.al.,Pianta molecolare, 2018
4.Storia demografica
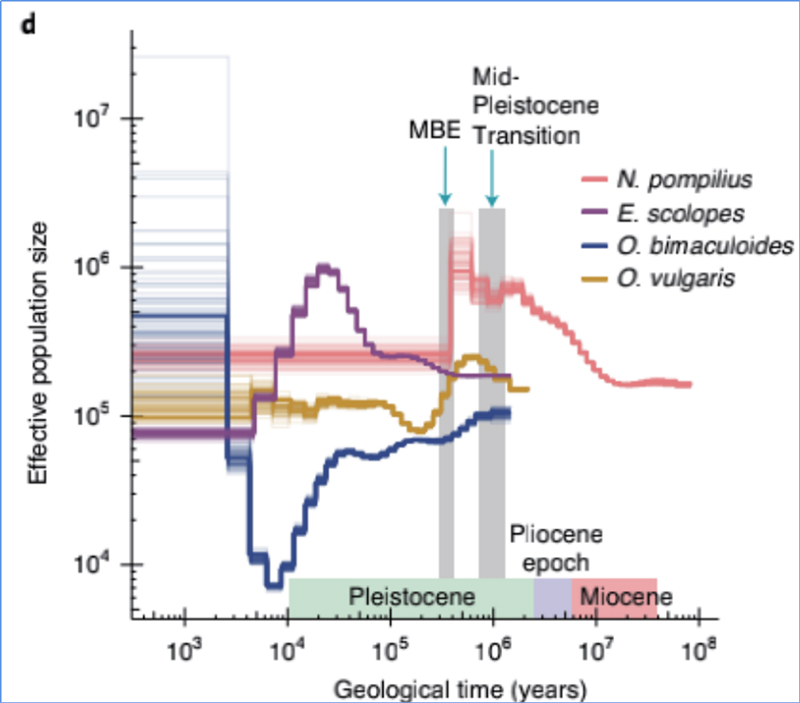
Zhang, et.al.,Ecologia&Evoluzione della Natura, 2021
5.Tempo di divergenza
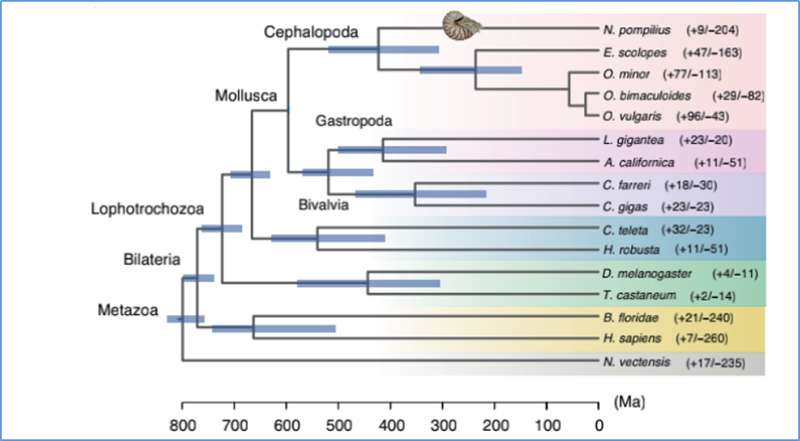
Zhang, et.al.,Ecologia&Evoluzione della Natura, 2021
Caso BMK
Una mappa della variazione genomica fornisce informazioni sulle basi genetiche della selezione del cavolo cinese primaverile (Brassica rapa ssp. Pekinensis)
Pubblicato: Pianta molecolare, 2018
Strategia di sequenziamento:
Risequenziamento: profondità di sequenziamento: 10×
Risultati chiave
In questo studio, 194 cavoli cinesi sono stati elaborati per il risequenziamento con una profondità media di 10×, che ha prodotto 1.208.499 SNP e 416.070 InDel.L'analisi filogenetica su queste 194 linee ha dimostrato che queste linee possono essere suddivise in tre ecotipi, primaverile, estivo e autunnale.Inoltre, la struttura della popolazione e l'analisi PCA hanno indicato che il cavolo cinese primaverile proveniva da un cavolo autunnale nello Shandong, in Cina.Questi furono successivamente introdotti in Corea e Giappone, incrociati con linee locali e alcune varietà a maturazione tardiva furono introdotte in Cina e infine divennero cavoli cinesi primaverili.
La scansione dell'intero genoma sui cavoli cinesi primaverili e autunnali durante la selezione ha rivelato 23 loci genomici che hanno subito una forte selezione, due dei quali erano sovrapposti con una regione di controllo del tempo di bullonamento basata sulla mappatura QTL.Si è scoperto che queste due regioni contengono geni chiave che regolano la fioritura, BrVIN3.1 e BrFLC1.È stato ulteriormente confermato che questi due geni sono coinvolti nel tempo di innesto mediante studi sul trascrittoma ed esperimenti transgenici.
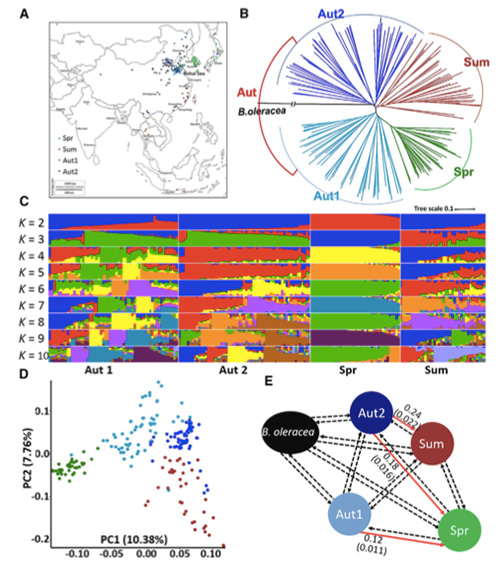 Analisi della struttura della popolazione sui cavoli cinesi | 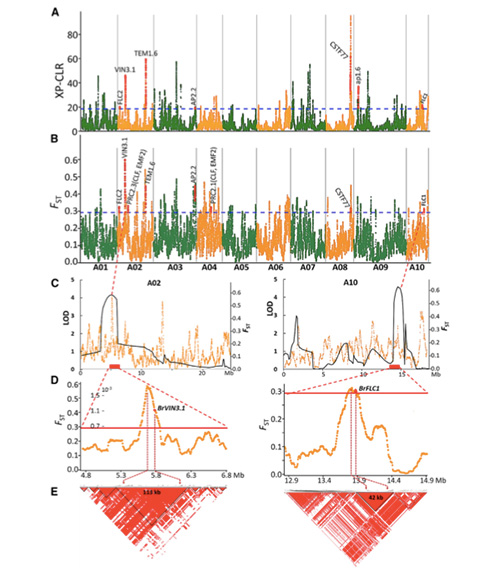 Informazioni genetiche sulla selezione del cavolo cinese |
Tongbing, et al."Una mappa di variazione genomica fornisce approfondimenti sulla base genetica della selezione del cavolo cinese primaverile (Brassica rapa ssp.pekinensis)."Piante molecolari,11(2018):1360-1376.





