

Genomica comparativa






Vantaggi del servizio
● Pacchetto di analisi completo, contenente le otto analisi più comunemente richieste
● Elevata affidabilità nell'analisi con interpretazione dettagliata e facilmente comprensibile dei risultati
● Dati ben progettati e pronti per la pubblicazione
● Un team di bioinformatica altamente qualificato soddisfa diverse richieste di analisi personalizzate
● Tempi di consegna più brevi con maggiore precisione nell'analisi
● Esperienza abbondante con oltre 90 casi di successo con un fattore di impatto cumulativo pubblicato di oltre 900
Specifiche del servizio
| Tempo di consegna stimato | Numero di specie | Analisi |
| 30 giorni lavorativi | 6 - 12 | Clustering della famiglia genetica Espansione e contrazione della famiglia genetica Costruzione di alberi filogenetici Stima del tempo di divergenza (è richiesta la calibrazione fossile) Tempo di inserimento LTR (Per impianti) Duplicazione dell'intero genoma (per le piante) Pressione selettiva Analisi della sintesi |
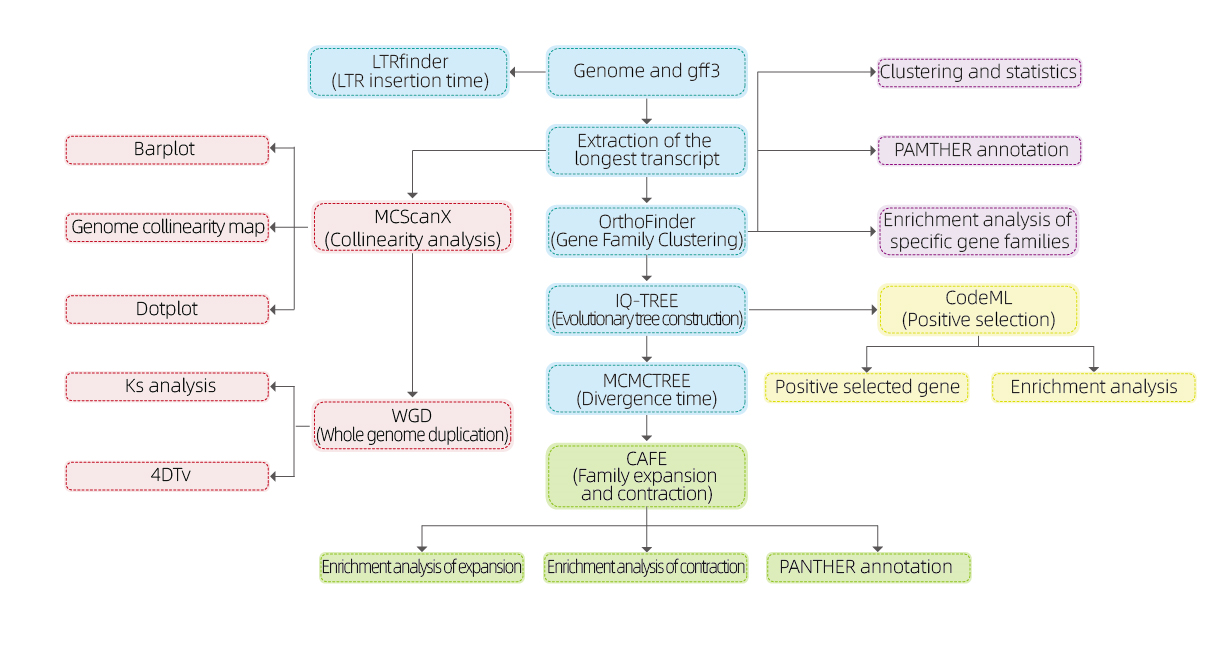
Analisi bioinformatiche
● Famiglia genetica
● Filogenetica
● Tempo di divergenza
● Pressione selettiva
● Analisi della sintesi
Requisiti e consegna del campione
Requisiti del campione:
Tessuto o DNA per il sequenziamento e l'assemblaggio del genoma
Per i tessuti
| Specie | Tessuto | Sondaggio | PacBioCCS |
| Animale | Tessuto viscerale | 0,5 ~ 1 g | ≥ 3,5 g |
| Tessuto muscolare | |||
| ≥ 5,0 g | |||
| ≥ 5,0 ml | |||
| Sangue di mammiferi | |||
| ≥ 0,5 ml | |||
| Sangue di pollame/pesce | |||
| Pianta | Foglia fresca | 1 ~ 2 g | ≥ 5,0 g |
| Petalo/stelo | 1 ~ 2 g | ≥ 10,0 g | |
| Radice/seme | 1 ~ 2 g | ≥ 20,0 g | |
| Celle | Cellula in coltura | - | ≥ 1×108 |
Dati
File di sequenza del genoma (.fasta) e file di annotazioni (.gff3) di specie strettamente correlate
Flusso di lavoro del servizio

Progettazione dell'esperimento

Consegna del campione

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita
*I risultati dimostrativi mostrati qui provengono tutti da genomi pubblicati con Biomarker Technologies
1. Stima del tempo di inserimento LTR: la figura mostra una distribuzione bimodale unica nei tempi di inserimento degli LTR-RT nel genoma della segale Weining, rispetto ad altre specie.Il picco più recente è apparso circa 0,5 milioni di anni fa.
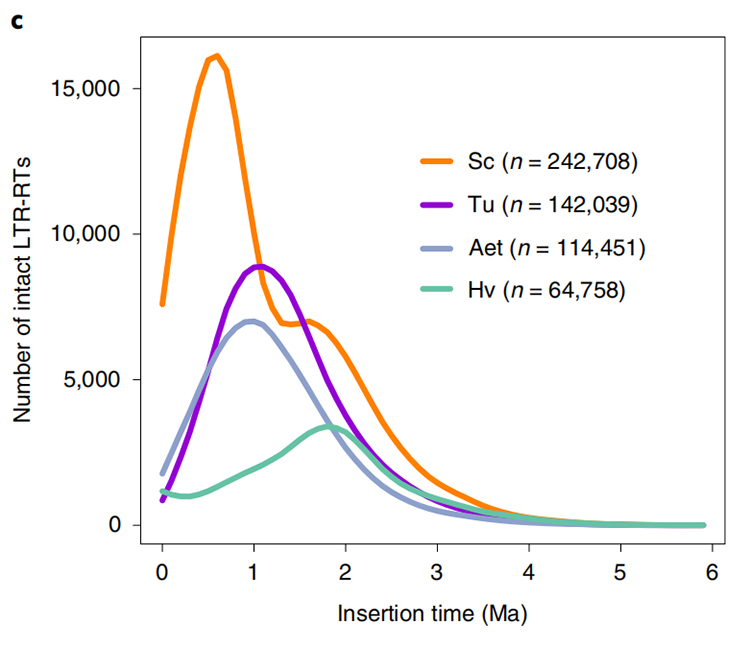
Li Guang et al.,Genetica della natura, 2021
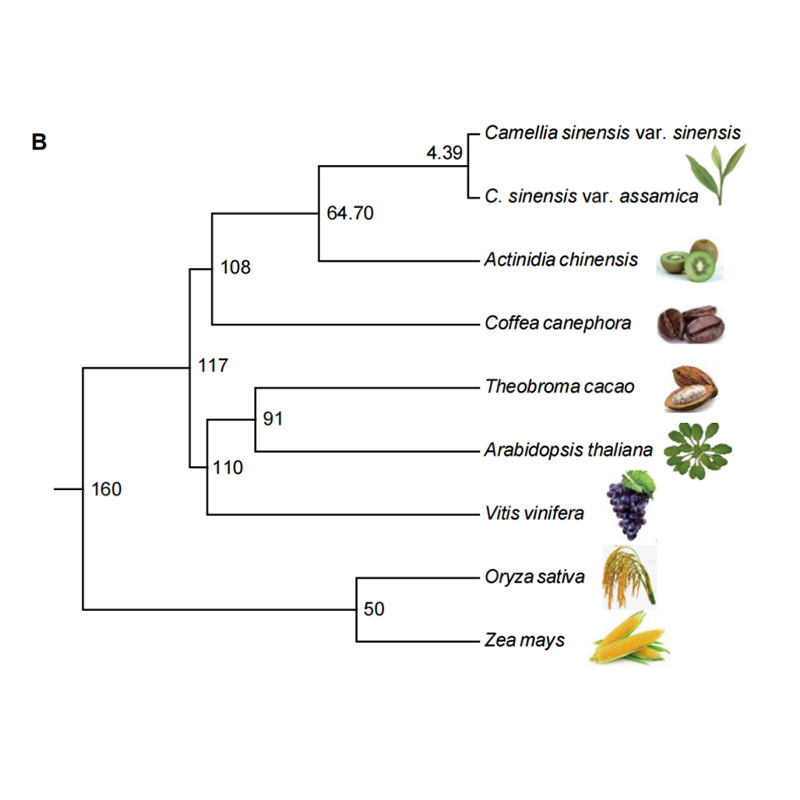
2. Analisi filogenetica e della famiglia genetica del chayote (Sechium edule): analizzando il chayote e le altre 13 specie correlate nella famiglia genetica, si è scoperto che il chayote è strettamente correlato alla zucca serpente (Trichosanthes anguina).Chayote derivato dalla zucca serpente in circa 27-45 Mya e la duplicazione dell'intero genoma (WGD) è stata osservata nel chayote in 25 ± 4 Mya, che è il terzo evento WGD nelle cucuibitaceae.
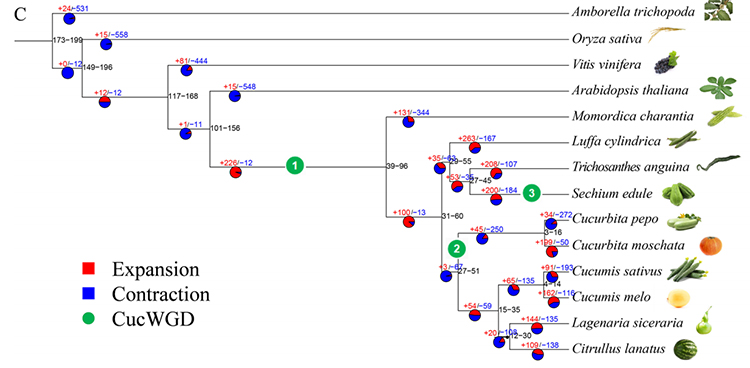
Fu A et al.,Ricerca sull'orticoltura, 2021
3. Analisi di sintesi: alcuni geni correlati ai fitormoni nello sviluppo del frutto sono stati trovati nel chayote, nella zucca serpente e nella zucca.La correlazione tra chayote e zucca è leggermente superiore a quella tra chayote e zucca serpente.
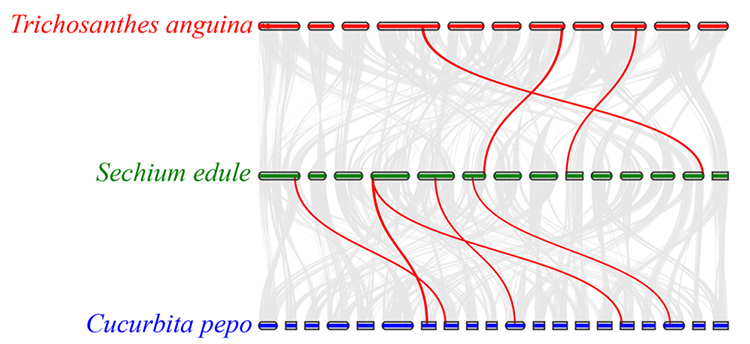
Fu A et al.,Ricerca sull'orticoltura, 2021
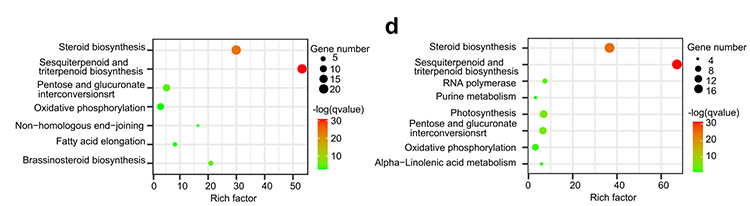
4. Analisi della famiglia genetica: l'arricchimento di KEGG sull'espansione e la contrazione della famiglia genetica nei genomi di G.thurberi e G.davidsonii ha dimostrato che i geni correlati alla biosintesi degli steroidi e alla biosintesi dei brassinosteroidi erano espansi.
Yang Z et al.,Biologia BMC, 2021
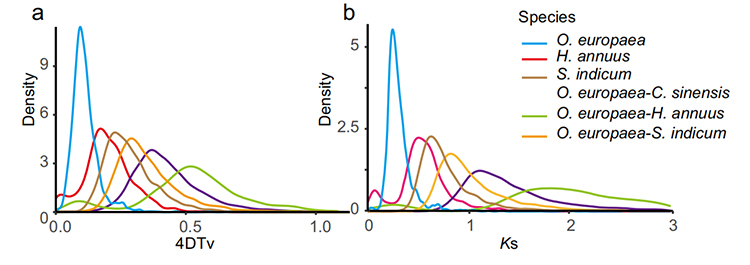
5. Analisi della duplicazione dell'intero genoma: l'analisi della distribuzione 4DTV e Ks ha mostrato l'evento di duplicazione dell'intero genoma.I picchi di intraspecie hanno mostrato eventi di duplicazione.Picchi di interspecie hanno mostrato eventi di speciazione.L'analisi ha indicato che, rispetto alle altre tre specie strettamente imparentate, O. europaea ha subito più recentemente una duplicazione genetica su larga scala.
Rao G et al.,Ricerca sull'orticoltura, 2021
Caso BMK
Rosa senza spine: intuizioni genomiche legate all'adattamento all'umidità
Pubblicato: Rassegna scientifica nazionale, 2021
Strategia di sequenziamento:
«BasyeSenza spine' (R.Wichurainan) genoma:
ca.93 X PacBio + ca.90 X nanopori + 267 X Illumina
Risultati chiave
1. Il genoma di R.wichuraiana di alta qualità è stato costruito utilizzando tecniche di sequenziamento a lettura lunga, che producono un assemblaggio di 530,07 Mb (la dimensione stimata del genoma era di circa 525,9 Mb mediante citometria a flusso e 525,5 mediante sondaggio del genoma; l'eterozigosità era di circa 1,03%).Il punteggio stimato da BUSCO era del 93,9%.Confrontando con "Old blush" (haploOB), la qualità e la completezza di questo genoma sono state confermate dall'accuratezza della base singola e dall'indice di assemblaggio LTR (LAI = 20,03).Il genoma di R.wichuraiana contiene 32.674 geni codificanti proteine.
2. L'analisi congiunta multi-omica, consistente in genomica comparativa, trascrittomica e analisi QTL della popolazione genetica, ha rivelato la speciazione cruciale tra R. wichuraiana e Rosa chinensis.Inoltre, è probabile che la variazione di espressione di geni correlati nel QTL sia associata al modello di puntura del gambo.
L'analisi genomica comparativa tra Basye's Thornless e Rosa chinensis, inclusa l'analisi della sintesi, il cluster della famiglia genetica, l'analisi dell'espansione e della contrazione, ha rivelato un gran numero di variazioni correlate ai tratti cruciali delle rose.L'espansione unica nella famiglia dei geni NAC e FAR1/FRS era molto probabilmente associata alla resistenza alla macchia nera.
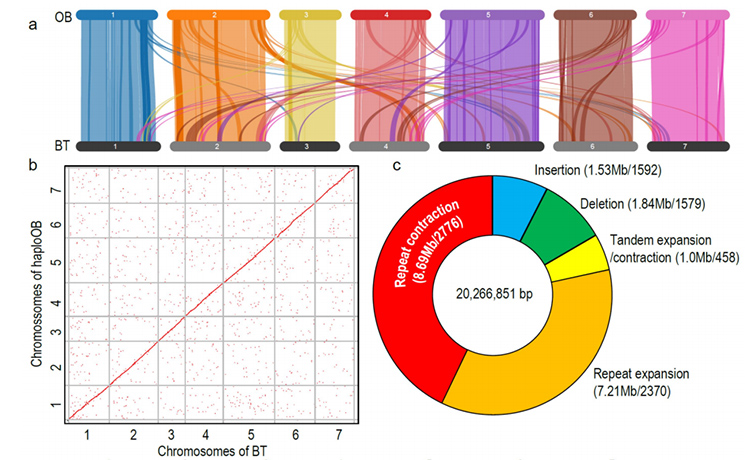

Analisi genomica comparativa tra genomi BT e haploOB.
Zhong, M., et al.“Rosa senza spine: approfondimenti genomici legati all’adattamento all’umidità”Rassegna scientifica nazionale, 2021;, nwab092.





