

Analisi dei segreganti in bulk






Vantaggi del servizio
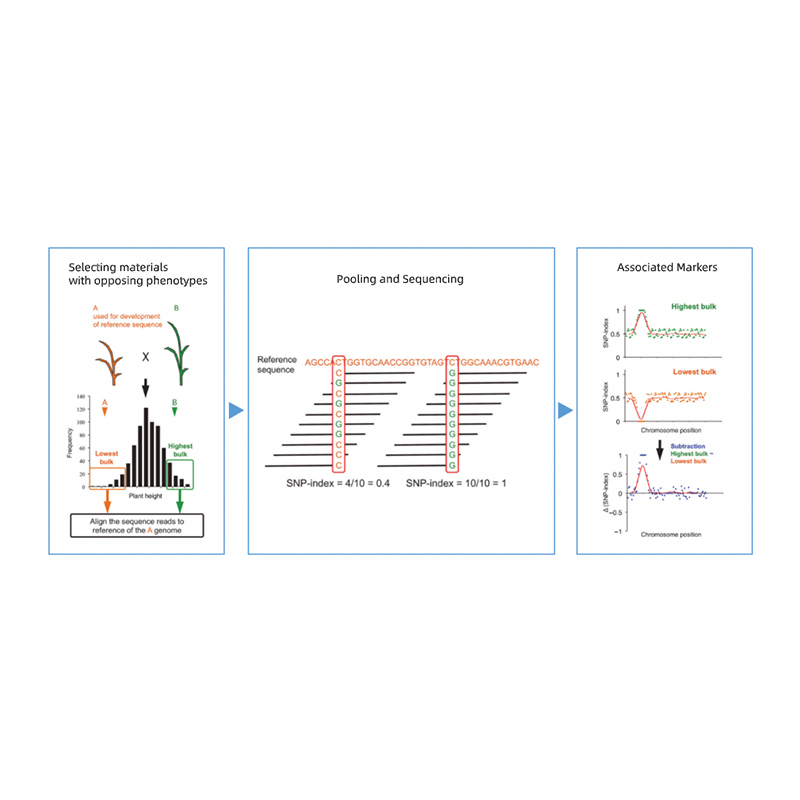
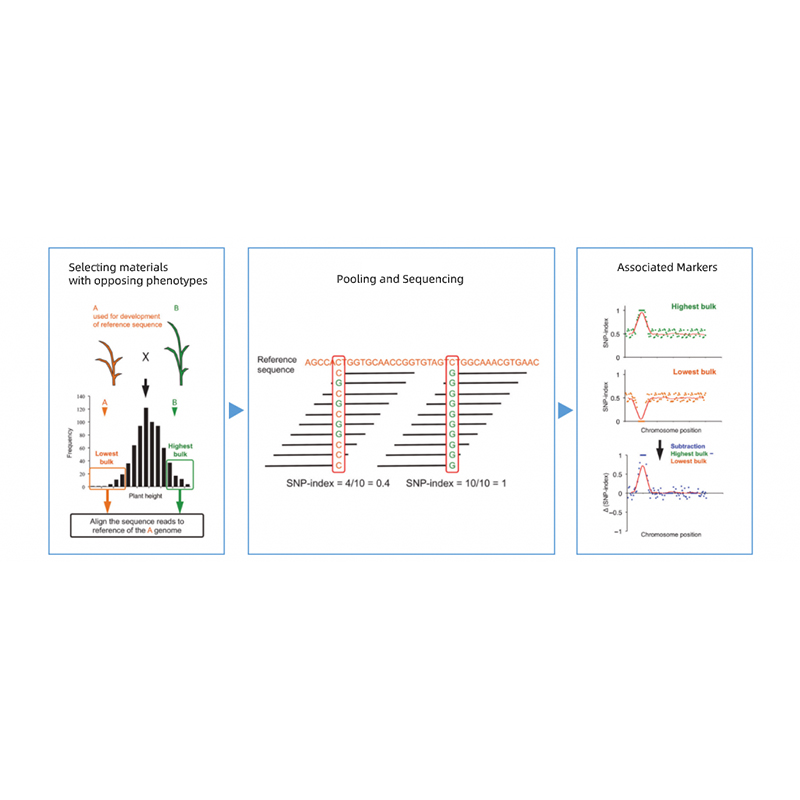
Takagi et al., Il diario delle piante, 2013
● Localizzazione accurata: miscelazione di gruppi da 30+30 a 200+200 individui per ridurre al minimo il rumore di fondo;previsione della regione candidata basata su mutanti non sinonimi.
● Analisi completa: annotazione approfondita della funzione del gene candidato, inclusi NR, SwissProt, GO, KEGG, COG, KOG, ecc.
● Tempi di consegna più rapidi: localizzazione rapida del gene entro 45 giorni lavorativi.
● Vasta esperienza: BMK ha contribuito alla localizzazione di migliaia di tratti, coprendo diverse specie come colture, prodotti acquatici, foreste, fiori, frutti, ecc.
Specifiche del servizio
Popolazione:
Segregazione della progenie di genitori con fenotipi opposti.
es. progenie F2, Backcrossing (BC), linea inbred ricombinante (RIL)
Piscina di miscelazione
Per i tratti qualitativi: da 30 a 50 individui (minimo 20)/gruppo
Per tratti quantitativi: dal 5% al 10% degli individui con uno dei due fenotipi estremi nell'intera popolazione (minimo 30+30).
Profondità di sequenziamento consigliata
Almeno 20X/genitore e 1X/prole individuale (ad esempio, per un pool di discendenti di 30+30 individui, la profondità di sequenziamento sarà 30X per massa)
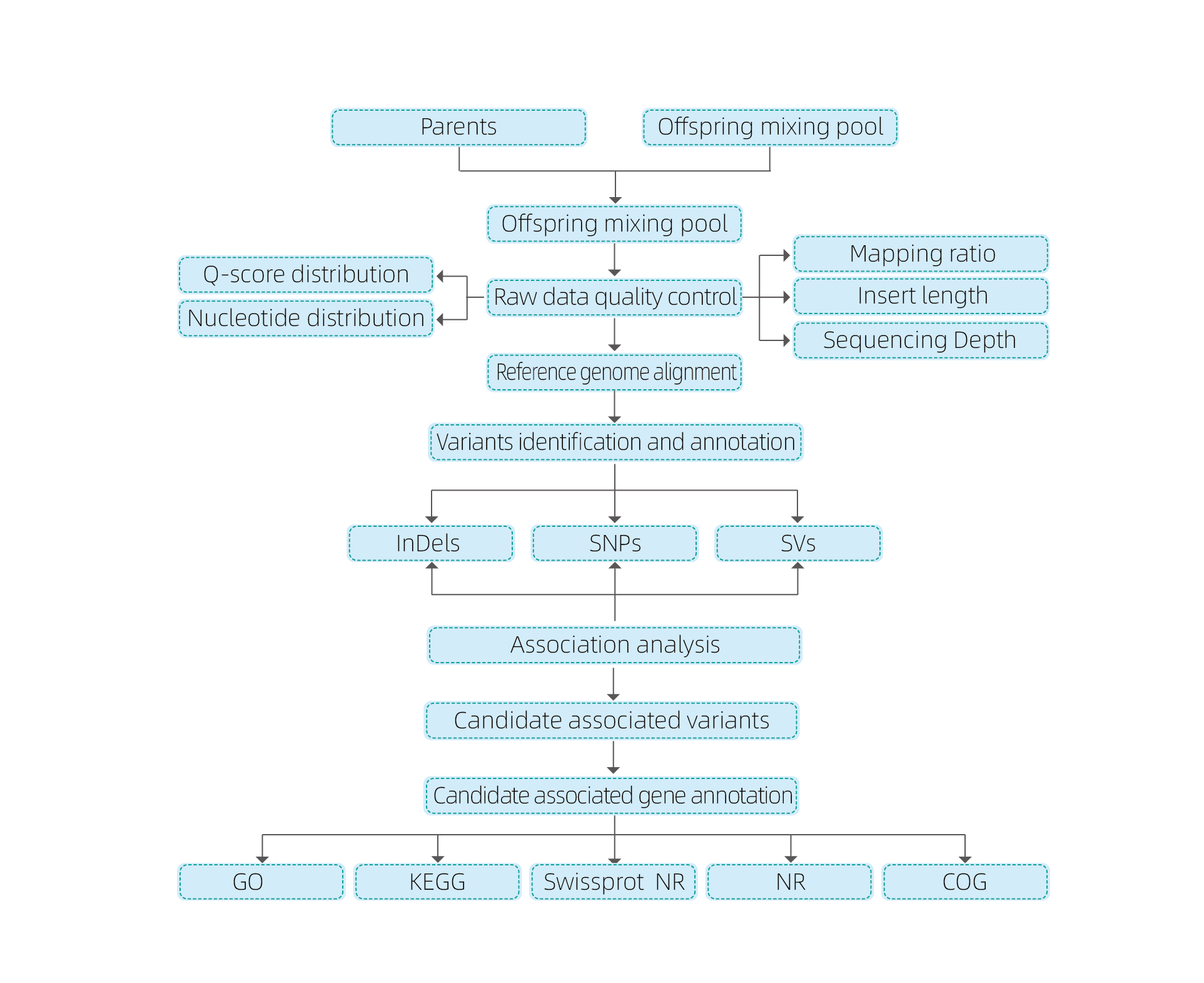
Analisi bioinformatiche
● Risequenziamento dell'intero genoma
● Elaborazione dei dati
● Chiamate SNP/Indel
● Screening delle regioni candidate
● Annotazione della funzione del gene candidato
Requisiti e consegna del campione
Requisiti del campione:
Nucleotidi:
| campione di gDNA | Campione di tessuto |
| Concentrazione: ≥30 ng/μl | Piante: 1-2 g |
| Quantità: ≥2 μg (Volume ≥15 μl) | Animali: 0,5-1 g |
| Purezza: OD260/280= 1,6-2,5 | Sangue intero: 1,5 ml |
Flusso di lavoro del servizio

Progettazione dell'esperimento

Consegna del campione

Estrazione dell'RNA

Costruzione della biblioteca

Sequenziamento

Analisi dei dati

Servizi post-vendita
1.Analisi dell'associazione basata sulla distanza euclidea (ED) per identificare la regione candidata.Nella figura seguente
Asse X: numero di cromosomi;Ogni punto rappresenta un valore ED di un SNP.La linea nera corrisponde al valore ED adattato.Un valore ED più elevato indica un'associazione più significativa tra il sito e il fenotipo.La linea tratteggiata rossa rappresenta la soglia di associazione significativa.
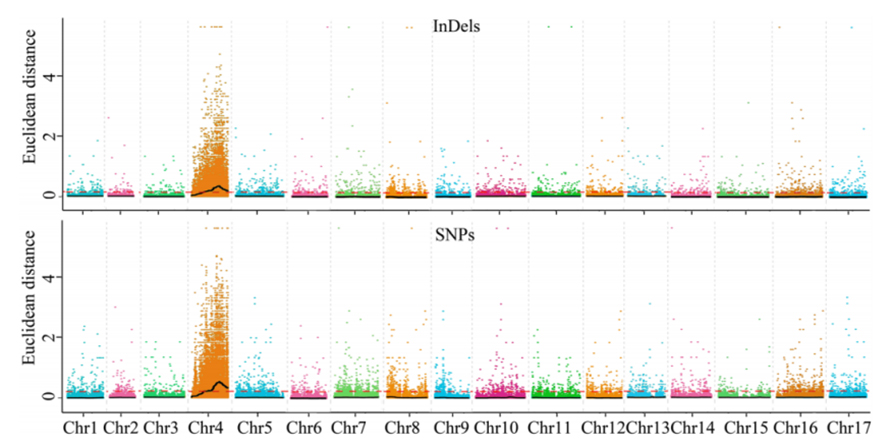
2.Analisi di associazione basata sull'assenza dell'indice SNP
Asse X: numero di cromosomi;Ogni punto rappresenta il valore dell'indice SNP.La linea nera rappresenta il valore dell'indice SNP adattato.Maggiore è il valore, più significativa è l'associazione.
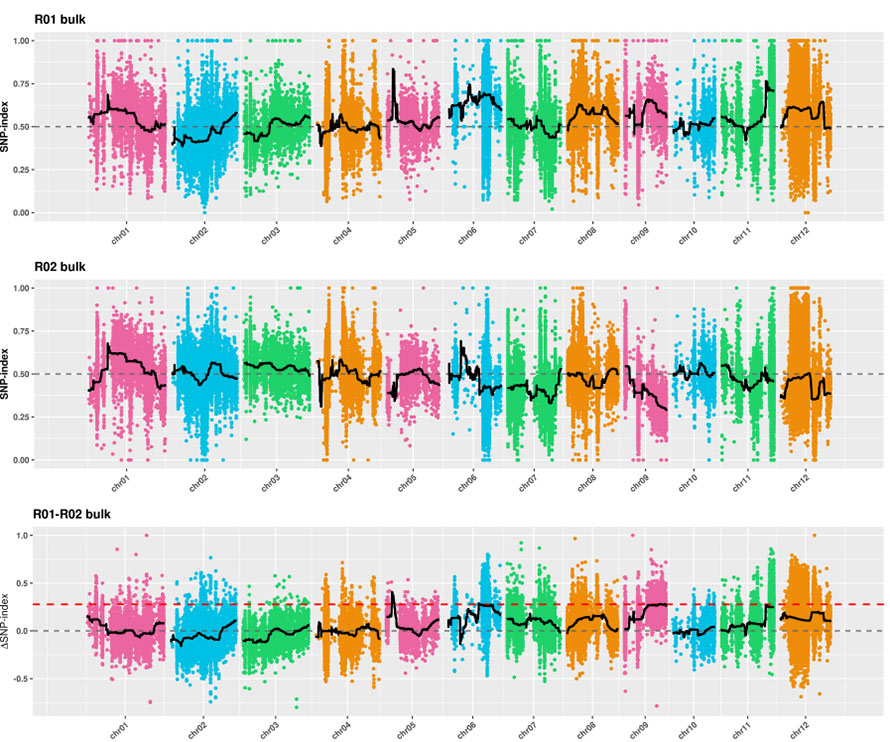
Caso BMK
Il locus del tratto quantitativo ad effetto maggiore Fnl7.1 codifica per una proteina abbondante nell'embriogenesi tardiva associata alla lunghezza del collo del frutto nel cetriolo
Pubblicato: Giornale delle biotecnologie vegetali, 2020
Strategia di sequenziamento:
Genitori (Jin5-508, YN): risequenziamento dell'intero genoma per 34× e 20×.
Pool di DNA (50 a collo lungo e 50 a collo corto): risequenziamento per 61× e 52×
Risultati chiave
In questo studio, la popolazione segregante (F2 e F2:3) è stata generata incrociando la linea di cetriolo a collo lungo Jin5-508 e YN a collo corto.Sono stati costruiti due pool di DNA da 50 individui dal collo estremamente lungo e da 50 individui dal collo estremamente corto.Il QTL con effetto maggiore è stato identificato su Chr07 mediante analisi BSA e mappatura QTL tradizionale.La regione candidata è stata ulteriormente ristretta mediante mappatura fine, quantificazione dell'espressione genica ed esperimenti transgenici, che hanno rivelato il gene chiave nel controllo della lunghezza del collo, CsFnl7.1.Inoltre, è stato scoperto che il polimorfismo nella regione del promotore CsFnl7.1 era associato all'espressione corrispondente.Ulteriori analisi filogenetiche hanno suggerito che è molto probabile che il locus Fnl7.1 abbia origine dall'India.
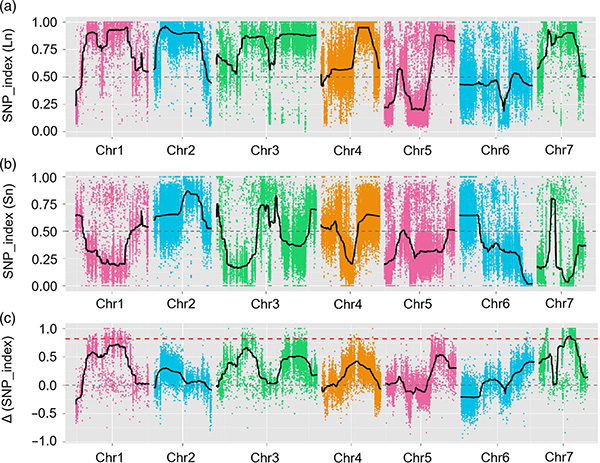 Mappatura QTL nell'analisi BSA per identificare la regione candidata associata alla lunghezza del collo del cetriolo | 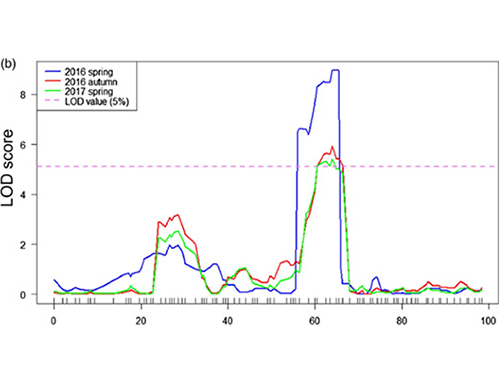 Profili LOD dei QTL della lunghezza del collo di cetriolo identificati su Chr07 |
Xu, X., et al."Il locus del tratto quantitativo ad effetto principale Fnl7.1 codifica per una proteina abbondante nell'embriogenesi tardiva associata alla lunghezza del collo del frutto nel cetriolo."Giornale delle biotecnologie vegetali 18.7(2020).





