

Tömeges szegregáns elemzés






Szolgáltatás előnyei
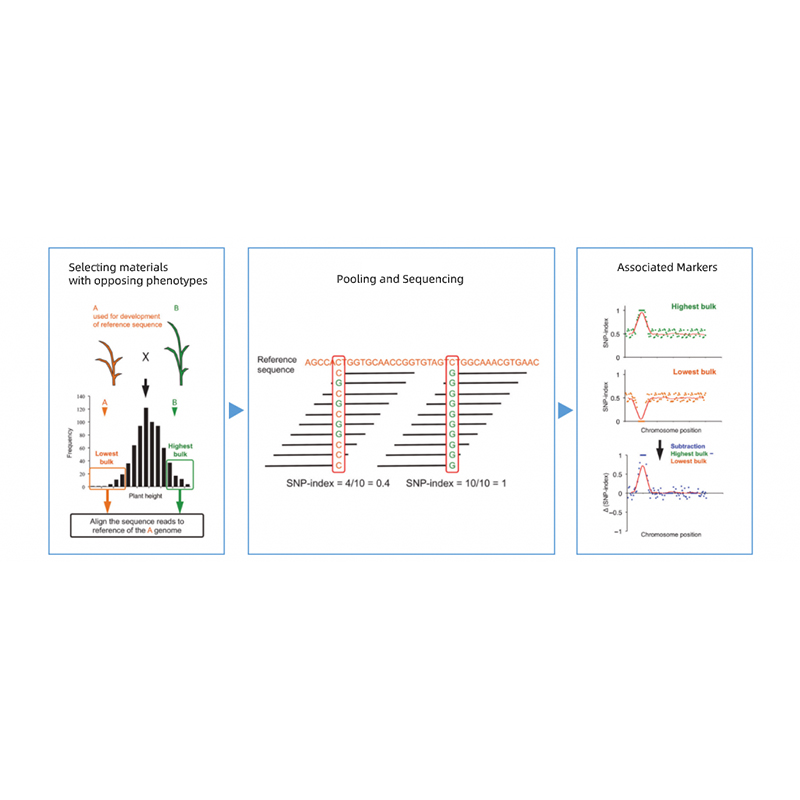
Takagi et al., The plant Journal, 2013
● Pontos lokalizáció: 30+30 és 200+200 egyed közötti tömegek keverése a háttérzaj minimalizálása érdekében;nem szinonim mutánsokon alapuló jelölt régió előrejelzés.
● Átfogó elemzés: mélyreható jelölt génfunkció annotáció, beleértve az NR, SwissProt, GO, KEGG, COG, KOG stb.
● Gyorsabb átfutási idő: Gyors génlokalizáció 45 munkanapon belül.
● Széleskörű tapasztalat: a BMK több ezer tulajdonság lokalizálásában járult hozzá, különféle fajok lefedésével, mint például a növények, vízi termékek, erdő, virágok, gyümölcsök stb.
Szolgáltatási specifikációk
Népesség:
Ellentétes fenotípusú szülők utódainak elkülönítése.
pl. F2 utód, Backcrossing (BC), Rekombináns beltenyésztett vonal (RIL)
Keverő medence
Minőségi tulajdonságok esetén: 30-50 egyed (minimum 20) / tömeg
Kvantitatív tratis esetén: a teljes populációban (minimum 30+30) az egyik szélsőséges fenotípusú egyed felső 5–10%-a.
Ajánlott szekvenálási mélység
Legalább 20X/szülő és 1X/utód egyed (pl. 30+30 egyedből álló ivadékkeverék esetén a szekvenálási mélység 30-szoros ömlesztve)
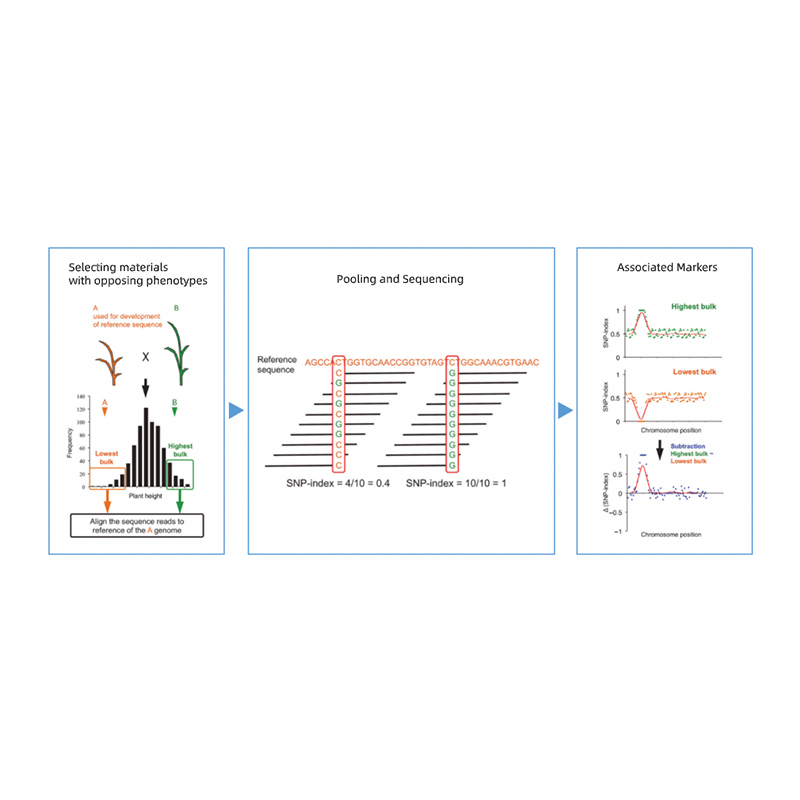
Bioinformatikai elemzések
● Teljes genom újraszekvenálása
● Adatfeldolgozás
● SNP/Indel hívás
● Jelölt régió szűrése
● Jelölt génfunkció annotáció
Mintakövetelmények és szállítás
Mintakövetelmények:
Nukleotidok:
| gDNS minta | Szövetminta |
| Koncentráció: ≥30 ng/μl | Növények: 1-2 g |
| Mennyiség: ≥2 μg (Térfogat ≥15 μl) | Állatok: 0,5-1 g |
| Tisztaság: OD260/280= 1,6-2,5 | Teljes vér: 1,5 ml |
Szerviz munkafolyamat

Kísérleti tervezés

Mintaszállítás

RNS extrakció

Könyvtárépítés

Sorrendezés

Adatelemzés

Értékesítés utáni szolgáltatások
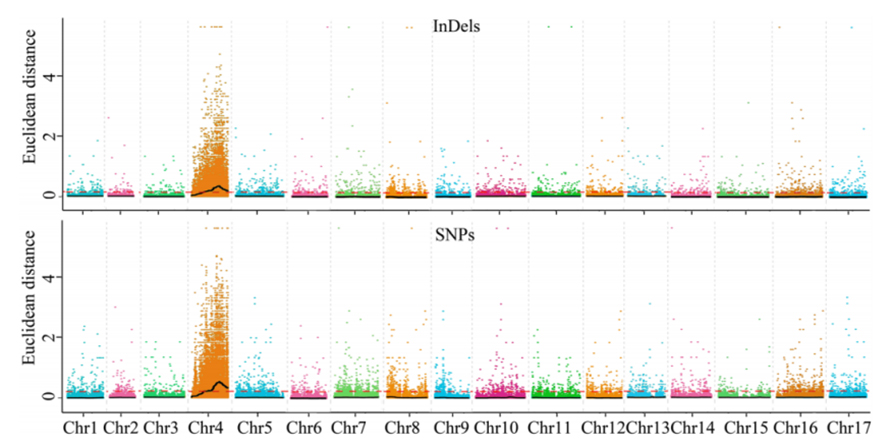
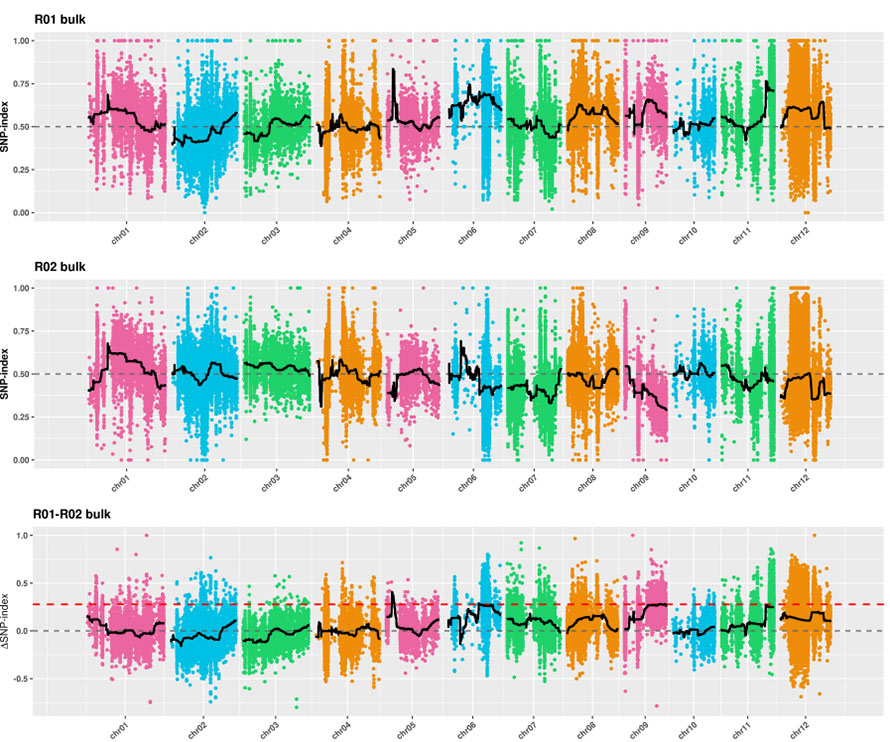
1. Euklideszi távolságon (ED) alapuló asszociációs elemzés a jelölt régió azonosításához.A következő ábrán
X-tengely: Kromoszómaszám;Minden pont egy SNP ED értékét jelenti.A fekete vonal az illesztett ED értéknek felel meg.A magasabb ED érték szignifikánsabb összefüggést jelez a hely és a fenotípus között.A piros szaggatott vonal a jelentős asszociáció küszöbét jelenti.
2. SNP-index nélküli asszociációs elemzés
X-tengely: Kromoszómaszám;Minden pont SNP-index értéket jelent.A fekete vonal az illesztett SNP-index értéket jelöli.Minél nagyobb az érték, annál jelentősebb az összefüggés.
BMK tok
Az Fnl7.1 fő hatású kvantitatív tulajdonságlókusz egy késői embriogenezisben gazdag fehérjét kódol, amely az uborka gyümölcsnyakhosszához kapcsolódik
Közzétett: Plant Biotechnology Journal, 2020
Szekvenálási stratégia:
Szülők (Jin5-508, YN): A teljes genom újraszekvenálása 34× és 20×.
DNS-készletek (50 hosszú nyakú és 50 rövid nyakú): Újraszekvenálás 61 × és 52 ×
Főbb eredmények
Ebben a vizsgálatban szegregáló populációt (F2 és F2:3) hoztak létre a hosszú nyakú uborkavonal Jin5-508 és a rövid nyakú YN keresztezésével.Két DNS-készletet építettek fel 50 extrém hosszú nyakú és 50 extrém rövid nyakú egyedből.A fő hatású QTL-t a Chr07-en azonosították BSA-analízissel és hagyományos QTL-leképezéssel.A jelölt régiót tovább szűkítettük finom térképezéssel, génexpresszió kvantifikációval és transzgenikus kísérletekkel, amelyek feltárták a nyakhossz szabályozásában kulcsfontosságú gént, a CsFnl7.1-et.Ezenkívül azt találtuk, hogy a CsFnl7.1 promóterrégió polimorfizmusa a megfelelő expresszióhoz kapcsolódik.További filogenetikai elemzések arra utaltak, hogy az Fnl7.1 lókusz nagy valószínűséggel Indiából származik.
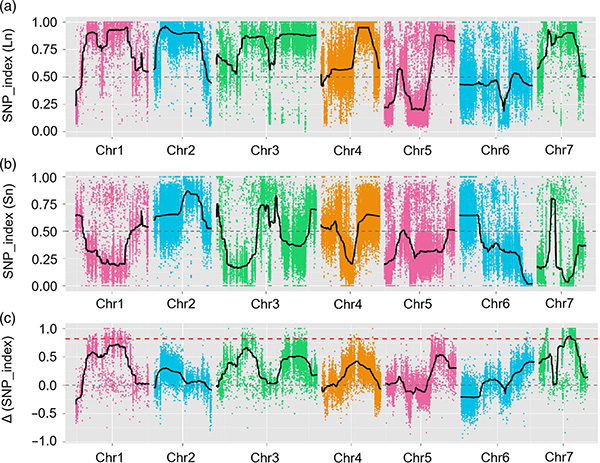 QTL-leképezés a BSA elemzésben az uborka nyakhosszához kapcsolódó jelölt régió azonosítására | 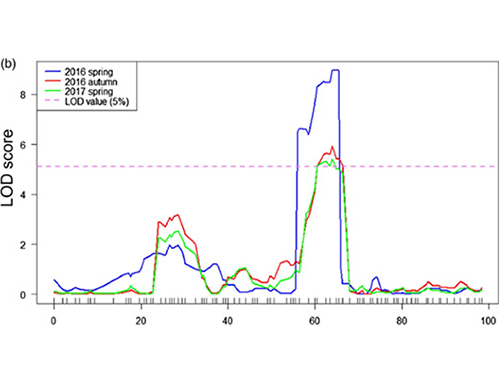 A Chr07-en azonosított uborka nyakhosszúságú QTL LOD profiljai |
Xu, X. et al."A fő hatást kiváltó Fnl7.1 kvantitatív tulajdonságlókusz egy késői embriogenezisben gazdag fehérjét kódol, amely az uborka gyümölcsnyakhosszához kapcsolódik."Plant Biotechnology Journal 18.7 (2020).





