

Asemblea xenómica baseada en Hi-C







Vantaxes do servizo
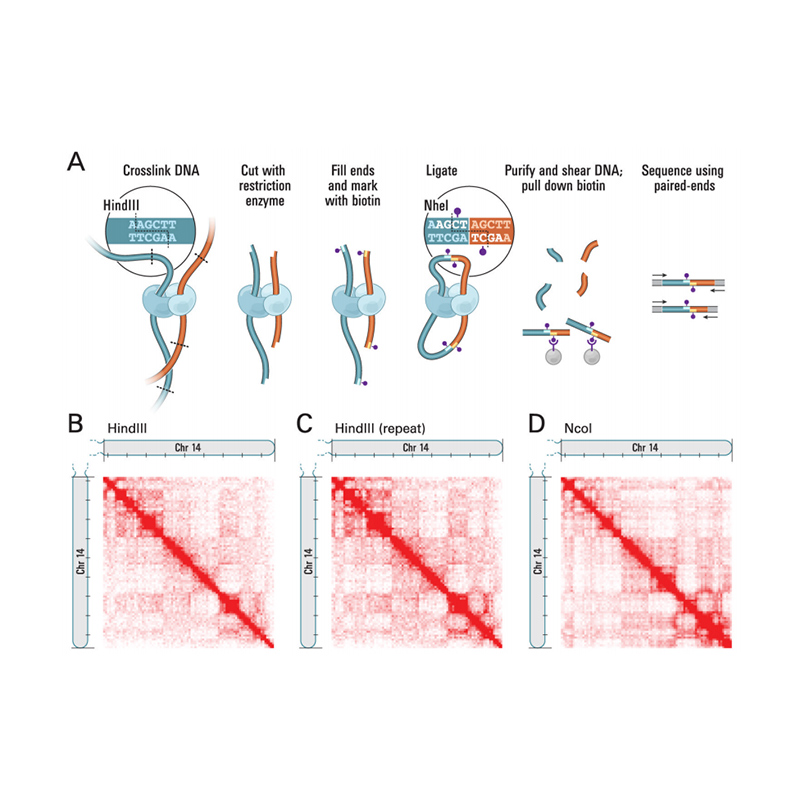
Visión xeral de Hi-C
(Lieberman-Aiden E et al.,Ciencia, 2009)
● Non é necesario construír poboación xenética para o ancoraxe contig;
● A maior densidade de marcadores leva a unha maior relación de ancoraxe de contigs por encima do 90%;
● Permite a avaliación e correccións de conxuntos de xenoma existentes;
● Menor tempo de resposta cunha maior precisión na montaxe do xenoma;
● Abundante experiencia con máis de 1000 bibliotecas Hi-C construídas para máis de 500 especies;
● Máis de 100 casos exitosos cun factor de impacto publicado acumulado superior a 760;
● Ensamblaxe do xenoma baseado en Hi-C para o xenoma poliploide, a taxa de ancoraxe do 100% conseguiuse no proxecto anterior;
● Patentes internas e dereitos de autor do software para experimentos Hi-C e análise de datos;
● Software de axuste de datos visualizados de desenvolvemento propio, permite o movemento manual de bloques, a marcha atrás, a revogación e a reposición.
Especificacións do servizo
|
Tipo de biblioteca
|
Plataforma | Lonxitude de lectura | Estratexia recomendada |
| Ola-C | Illumina NovaSeq | PE150 | ≥ 100X |
Análise bioinformática
● Control de calidade dos datos brutos
● Control de calidade da biblioteca Hi-C
● Ensamblaxe do xenoma baseado en Hi-C
● Avaliación posterior á montaxe
Requisitos de mostra e entrega
Requisitos de mostra:
| Animal | Fungo | Plantas
|
| Tecido conxelado: 1-2 g por biblioteca Celas: 1x 10^7 celas por biblioteca | Tecido conxelado: 1 g por biblioteca | Tecido conxelado: 1-2 g por biblioteca
|
| *Recomendámosche encarecidamente enviar polo menos 2 alícuotas (1 g cada unha) para o experimento Hi-C. | ||
Entrega de mostras recomendada
Recipiente: tubo de centrífuga de 2 ml (non se recomenda papel de aluminio)
Para a maioría das mostras, recomendamos non conservalas en etanol.
Etiquetado da mostra: as mostras deben estar claramente etiquetadas e idénticas ao formulario de información da mostra enviado.
Envío: Xeo seco: as mostras deben ser embaladas primeiro en bolsas e enterradas en xeo seco.
Fluxo de traballo do servizo

Deseño de experimentos

Entrega da mostra

extracción de ADN

Construción da biblioteca

Secuenciación

Análise de datos

Servizos posvenda
*Os resultados de demostración que se mostran aquí son todos de xenomas publicados con Biomarker Technologies
1.Mapa de calor de interacción Hi-C deCamptotheca acuminataxenoma.Como se mostra no mapa, a intensidade das interaccións está negativamente correlacionada coa distancia lineal, o que indica un conxunto de cromosomas moi precisos.(Proporción de ancoraxe: 96,03%)
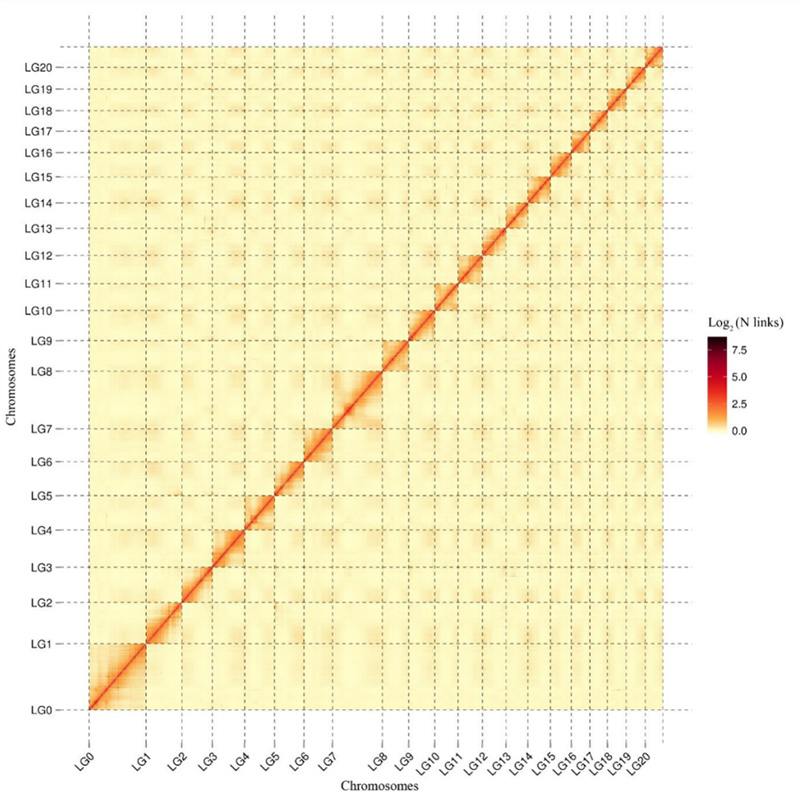
Kang M et al.,Comunicacións da natureza, 2021
2.Hi-C facilitou a validación das inversións entreGossypium hirsutumL. TM-1 A06 eG. arboreoChr06
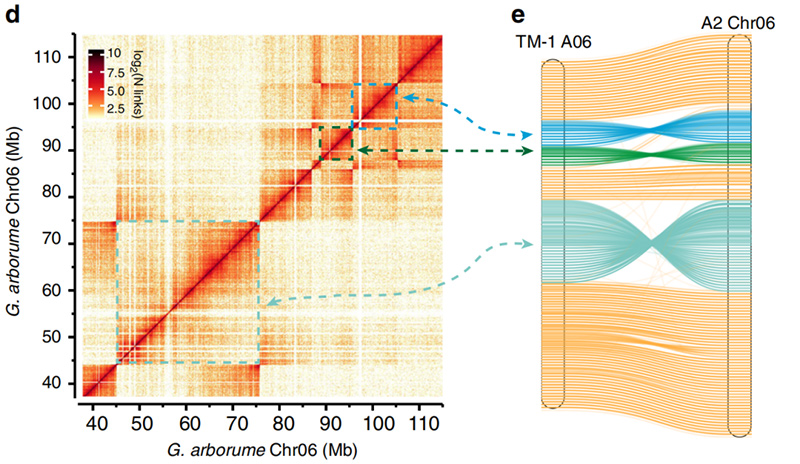
Yang Z et al.,Nature Communications, 2019
3.Ensamblaxe e diferenciación bialélica do xenoma da mandioca SC205.O mapa de calor Hi-C mostra unha clara división en cromosomas homólogos.
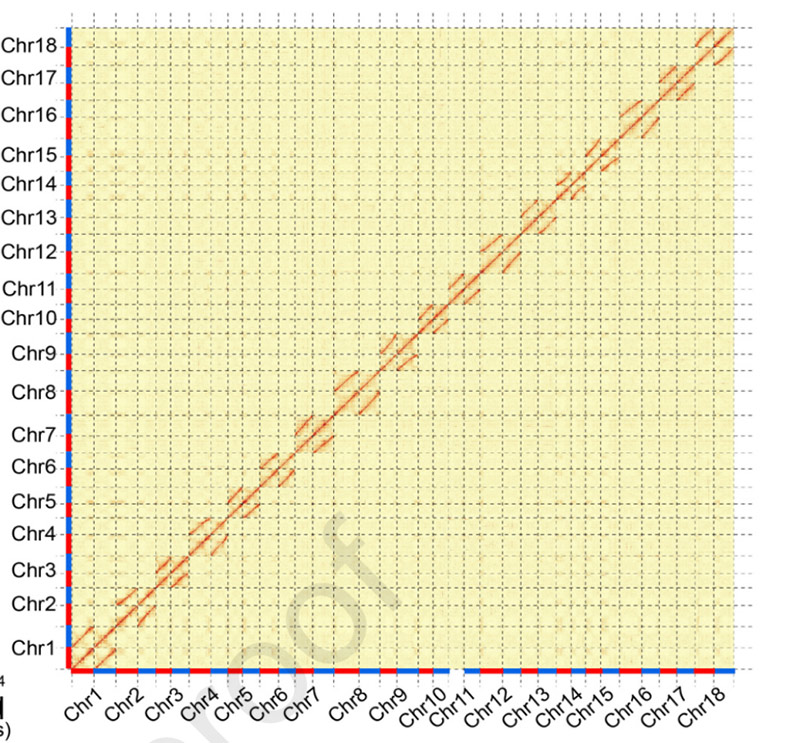
Hu W et al.,Planta Molecular, 2021
4.Mapa de calor Hi-C en conxunto de xenoma de dúas especies de Ficus:F.microcarpa(ratio de ancoraxe: 99,3%) eF.hispida (relación de ancoraxe: 99,7%)
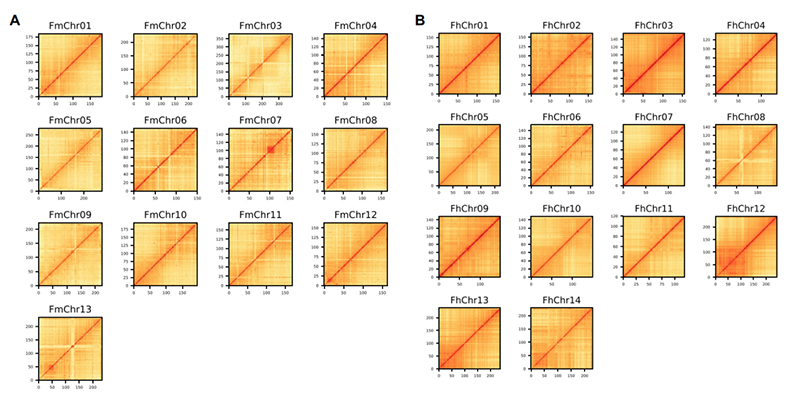
Zhang X et al.,Célula, 2020
Caso BMK
Os xenomas do Banyan Tree e da avespa polinizadora proporcionan información sobre a coevolución da avespa figueira
Publicado: Célula, 2020
Estratexia de secuenciación:
F. microcarpa xenoma: aprox.84 X PacBio RSII (36,87 Gb) + Hi-C (44 Gb)
F. hispidaxenoma: aprox.97 X PacBio RSII (36,12 Gb) + Hi-C (60 Gb)
Eupristina verticillataxenoma: aprox.170 X PacBio RSII (65 Gb)
Resultados clave
1.Construíronse dous xenomas de árbores de banyan e un xenoma de avespa polinizadora utilizando a secuenciación PacBio, Hi-C e mapa de enlace.
(1)F. microcarpaxenoma: estableceuse un conxunto de 426 Mb (97,7% do tamaño estimado do xenoma) cun contig N50 de 908 Kb, puntuación BUSCO do 95,6%.En total, secuencias de 423 Mb foron ancoradas a 13 cromosomas por Hi-C.A anotación do xenoma deu 29.416 xenes codificantes de proteínas.
(2)F. Hispidaxenoma: un conxunto de 360 Mb (97,3% do tamaño estimado do xenoma) produciuse cun contig N50 de 492 Kb e unha puntuación BUSCO do 97,4%.Un total de secuencias de 359 Mb foron ancoradas en 14 cromosomas por Hi-C e moi idénticas ao mapa de enlace de alta densidade.
(3)Eupristina verticillataxenoma: estableceuse un conxunto de 387 Mb (tamaño do xenoma estimado: 382 Mb) cun contig N50 de 3,1 Mb e unha puntuación BUSCO do 97,7%.
2.A análise xenómica comparada revelou un gran número de variacións de estrutura entre dousFicusxenomas, que proporcionaron un recurso xenético inestimable para estudos de evolución adaptativa.Este estudo, por primeira vez, proporcionou información sobre a coevolución da avespa figueira a nivel xenómico.
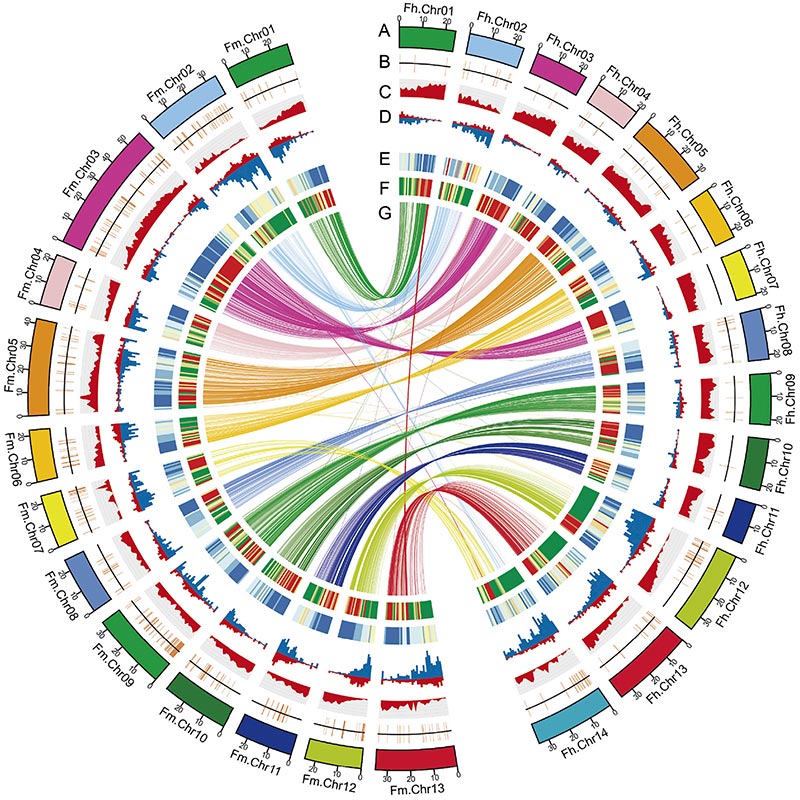 Diagrama de circos sobre as características xenómicas de dousFicusxenomas, incluíndo cromosomas, duplicacións segmentarias (SD), transposóns (LTR, TEs, TEs de ADN), expresión xénica e sintenía | 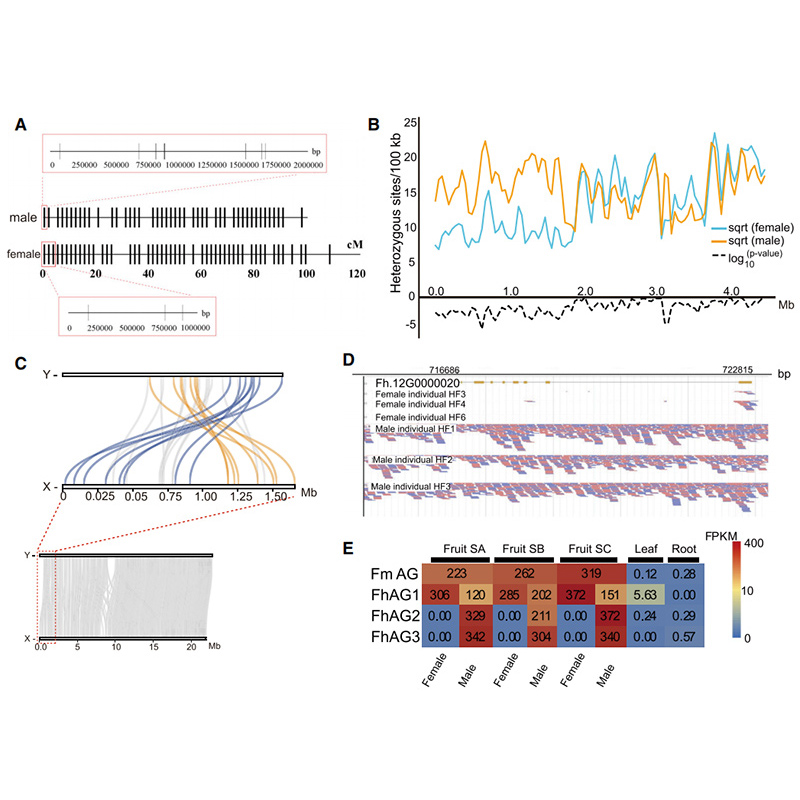 Identificación do cromosoma Y e xene candidato á determinación do sexo |
Zhang, X., et al."Os xenomas do Banyan Tree e a Polinizadora da avispa proporcionan información sobre a coevolución da figueira e avespa".Cela 183.4 (2020).





