

Xenética evolutiva








Vantaxes do servizo
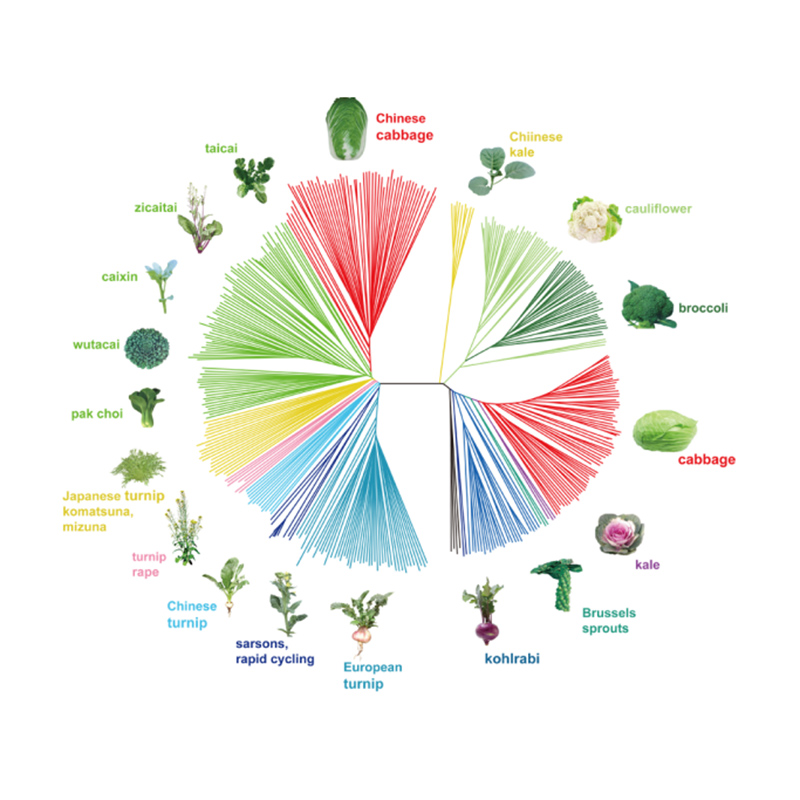
Takagi et al.,O diario das plantas, 2013
● Estimar o tempo e a velocidade de diverxencia das especies en función das variacións a nivel de nucleótidos e aminoácidos
● Revelación dunha relación filoxenética máis fiable entre especies cunha influencia minimizada da evolución converxente e da evolución paralela.
● Construír vínculos entre os cambios xenéticos e os fenotipos para descubrir xenes relacionados cos trazos
● Estimación da diversidade xenética, que reflicte o potencial evolutivo das especies
● Tempo de resposta máis rápido
● Ampla experiencia: BMK acumula unha experiencia masiva en proxectos relacionados coa poboación e a evolución durante máis de 12 anos, abarcando centos de especies, etc., e contribuíu en máis de 80 proxectos de alto nivel publicados en Nature Communications, Molecular Plants, Plant Biotechnology Journal, etc.
Especificacións do servizo
Materiais:
Normalmente, recoméndase polo menos tres subpoboacións (por exemplo, subespecies ou cepas).Cada subpoboación debe conter non menos de 10 individuos (plantas > 15, poden reducirse para especies raras).
Estratexia de secuenciación:
* WGS pódese empregar para especies con xenoma de referencia de alta calidade, mentres que SLAF-Seq é aplicable a especies con ou sen xenoma de referencia ou xenoma de referencia de mala calidade.
| Aplicable ao tamaño do xenoma | WGS | SLAF-Etiquetas (×10.000) |
| ≤ 500 Mb | 10×/individual | WGS é máis recomendable |
| 500 Mb - 1 Gb | 10 | |
| 1 Gb - 2 Gb | 20 | |
| ≥2 Gb | 30 |
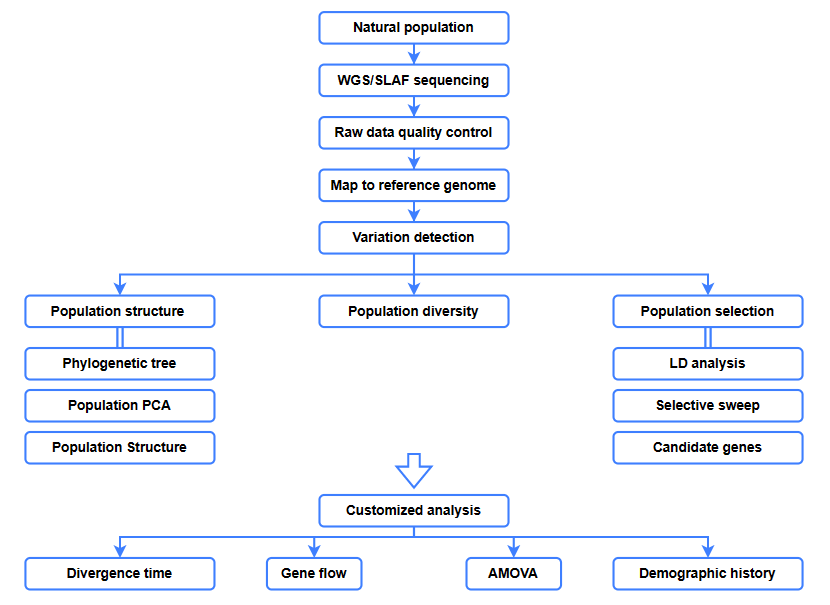
Análise bioinformática
● Análise evolutiva
● Varrido selectivo
● Fluxo xenético
● Historial demográfico
● Tempo de diverxencia
Requisitos de mostra e entrega
Requisitos de mostra:
| Especie | Tecido | WGS-NGS | SLAF |
| Animal
| Tecido visceral |
0,5 ~ 1 g
|
0,5 g
|
| Tecido muscular | |||
| Sangue de mamíferos | 1,5 ml
| 1,5 ml
| |
| Sangue de aves/peixes | |||
| Planta
| Folla Fresca | 1 ~ 2 g | 0,5 ~ 1 g |
| Pétalo/Talla | |||
| Raíz/Semente | |||
| Células | Célula cultivada |
| gDNA | Concentración | Cantidade (ug) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1,6-2,5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Fluxo de traballo do servizo

Deseño de experimentos

Entrega da mostra

Construción da biblioteca

Secuenciación

Análise de datos

Servizos posvenda
*Os resultados de demostración que se mostran aquí son todos de xenomas publicados con BMKGENE
1.A análise da evolución contén a construción da árbore filoxenética, a estrutura da poboación e a PCA baseada en variacións xenéticas.
A árbore filoxenética representa relacións taxonómicas e evolutivas entre especies cun antepasado común.
A PCA ten como obxectivo visualizar a proximidade entre subpoboacións.
A estrutura da poboación mostra a presenza de subpoboacións xeneticamente distintas en termos de frecuencias alelosas.
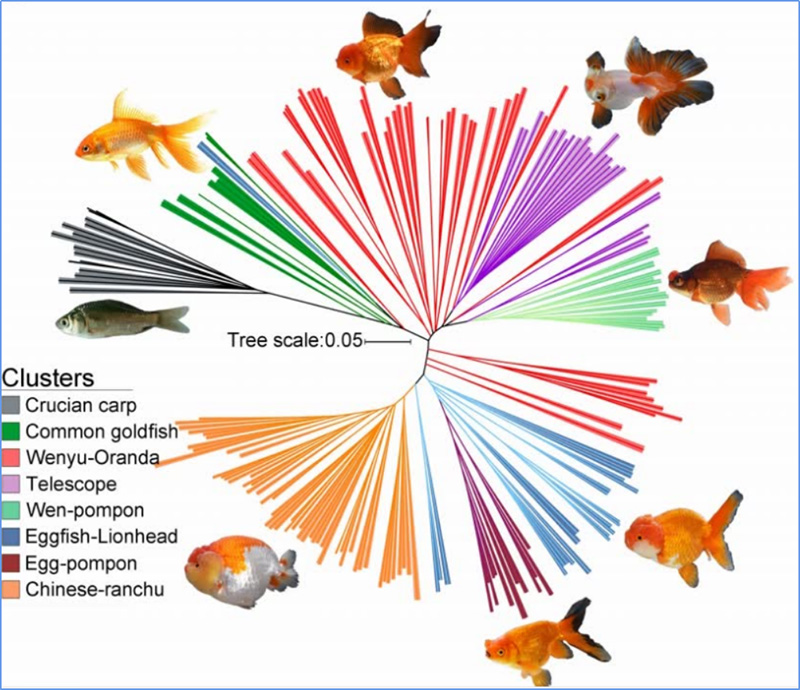
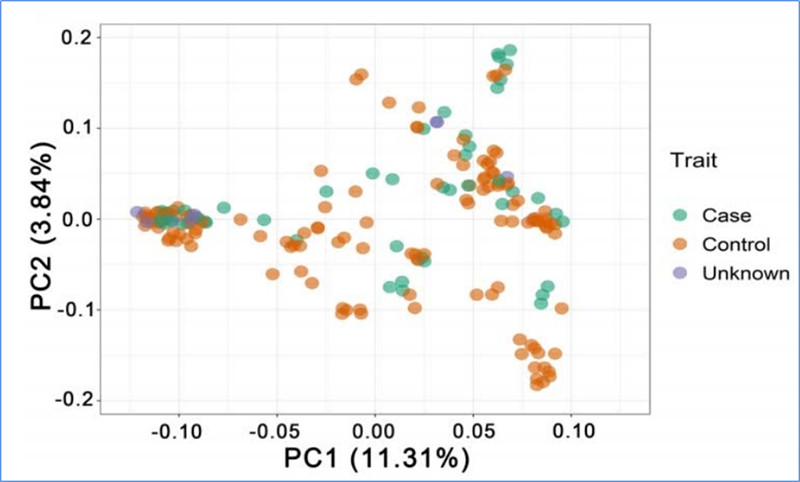
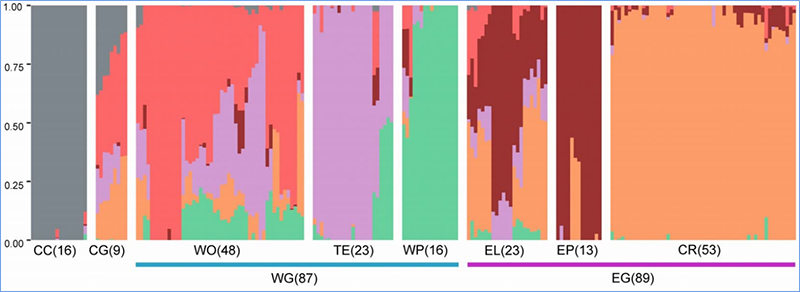
Chen, et.al.,PNAS, 2020
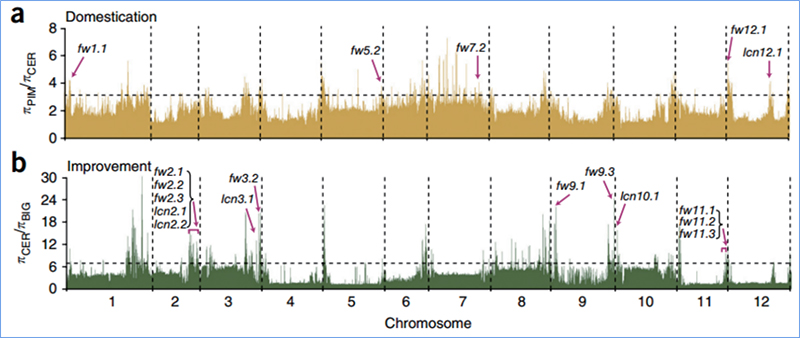
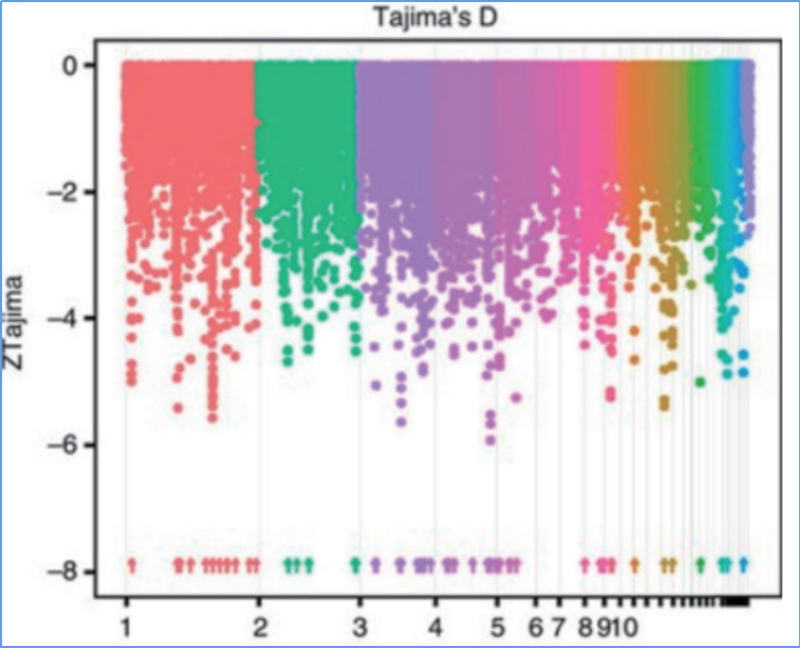
2.Varrido selectivo
O varrido selectivo refírese a un proceso polo cal se selecciona un sitio vantaxoso e aumentan as frecuencias de sitios neutros vinculados e diminúen as dos sitios sen vincular, o que resulta na redución do rexional.
A detección de todo o xenoma en rexións de varrido selectivo procédese calculando o índice xenético da poboación (π,Fst, D de Tajima) de todos os SNP dentro dunha xanela deslizante (100 Kb) nun determinado paso (10 Kb).
Diversidade de nucleótidos (π)
Tajima D
Índice de fixación (Fst)
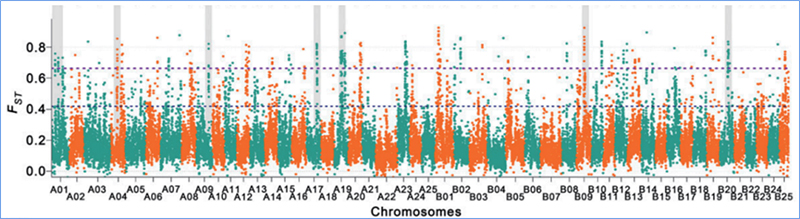
Wu, et.al.,Planta Molecular, 2018
3. Fluxo xenético
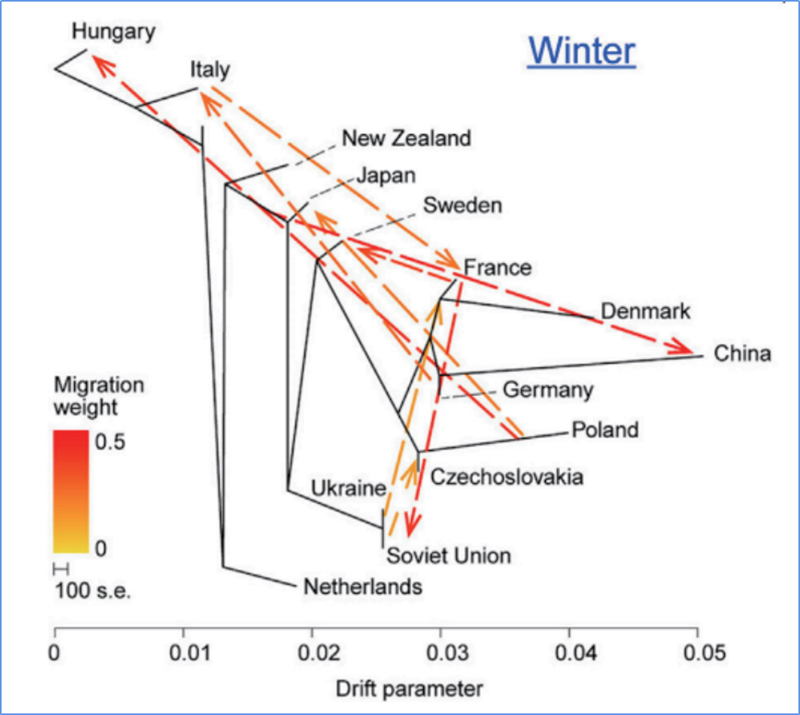
Wu, et.al.,Planta Molecular, 2018
4.Historia demográfica
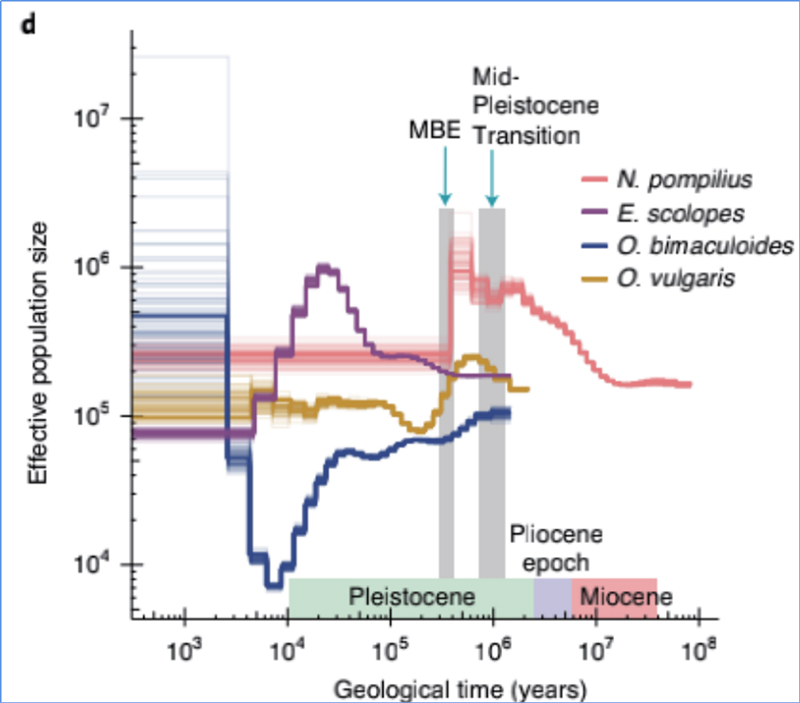
Zhang, et.al.,Natureza Ecoloxía e Evolución, 2021
5.Tempo de diverxencia
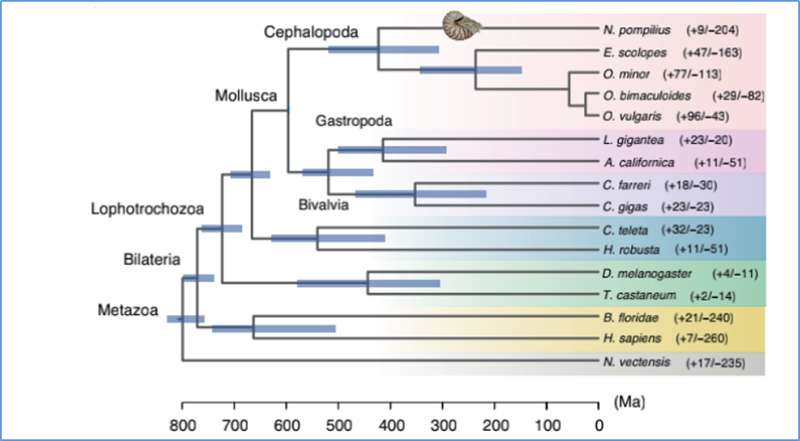
Zhang, et.al.,Natureza Ecoloxía e Evolución, 2021
Caso BMK
Un mapa de variacións xenómicas proporciona información sobre a base xenética da selección de repolo chino de primavera (Brassica rapa ssp. Pekinensis)
Publicado: Planta Molecular, 2018
Estratexia de secuenciación:
Resecuenciación: profundidade de secuenciación: 10×
Resultados clave
Neste estudo, procesáronse 194 repolos chinos para a súa secuenciación cunha profundidade media de 10 ×, o que deu 1.208.499 SNP e 416.070 InDels.A análise filoxenética destas 194 liñas demostrou que estas liñas pódense dividir en tres ecotipos, primavera, verán e outono.Ademais, a estrutura da poboación e a análise da PCA indicaron que a repolo chinesa de primavera orixinouse dun repolo de outono en Shandong, China.Estes foron introducidos posteriormente en Corea e Xapón, cruzados con liñas locais e algunhas variedades de parafusos tardíos foron introducidos de novo en China e finalmente convertéronse en repolo chino de primavera.
A exploración de todo o xenoma en repolos chinos de primavera e repolos de outono na selección revelou 23 loci xenómicos que pasaron por unha forte selección, dous dos cales se solaparon coa rexión de control do tempo de parafuso baseada na cartografía QTL.Descubriuse que estas dúas rexións conteñen xenes clave que regulan a floración, BrVIN3.1 e BrFLC1.Confirmouse ademais que estes dous xenes estaban implicados no tempo de perno mediante o estudo do transcriptoma e os experimentos transxénicos.
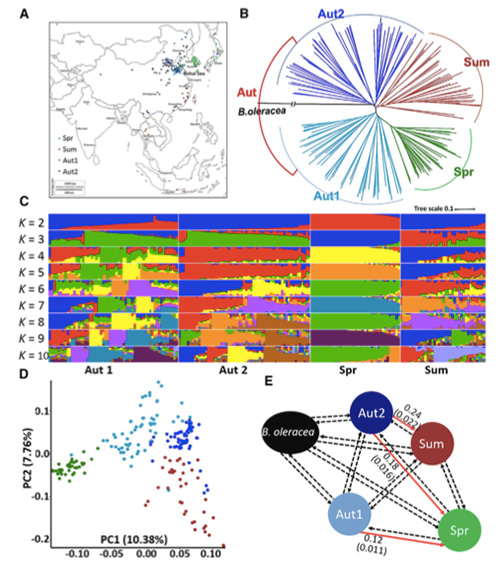 Análise da estrutura da poboación sobre repolos chineses | 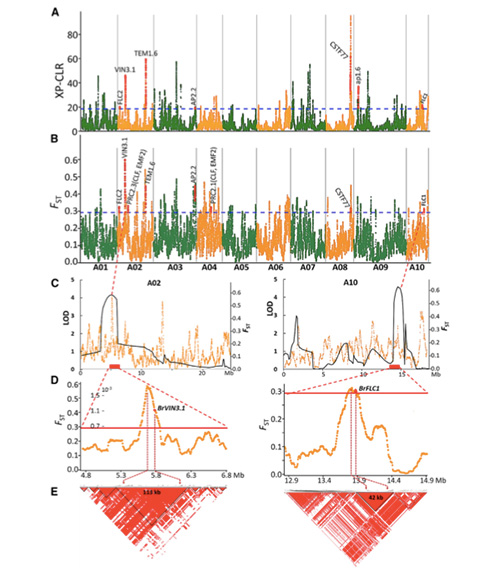 Información xenética sobre a selección de repolo chino |
Tongbing, et al."Un mapa de variacións xenómicas proporciona información sobre as bases xenéticas da selección de repolo chinés de primavera (Brassica rapa ssp.pekinensis)".plantas moleculares,11 (2018): 1360-1376.





