

Secuenciación de ARNm de lonxitude completa -PacBio





Vantaxes do servizo
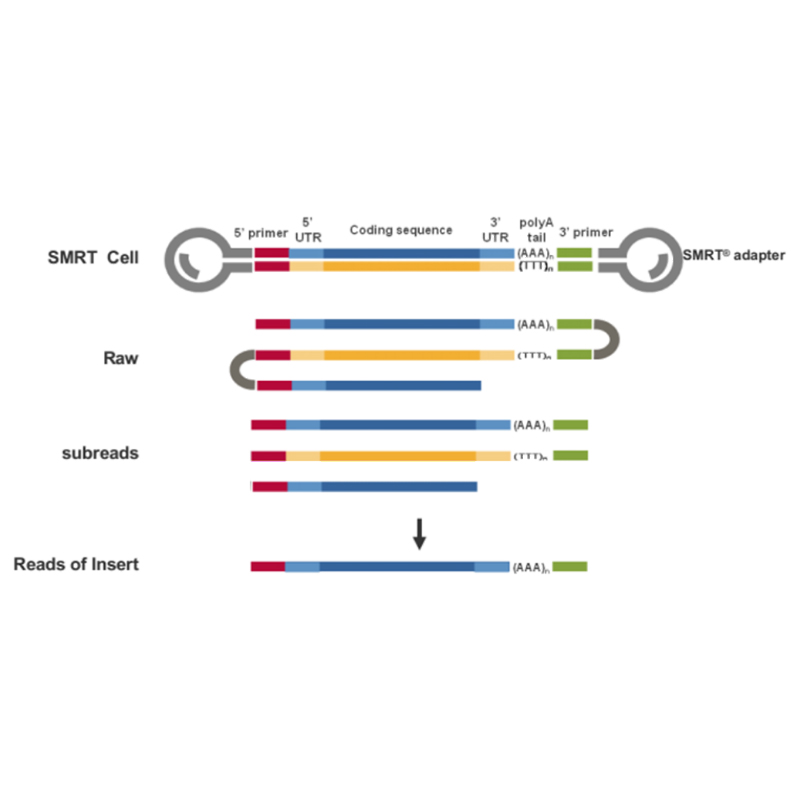
● Lectura directa da molécula de ADNc de lonxitude completa dende o extremo 3' ata o extremo 5'
● Resolución de nivel de forma iso na estrutura de secuencia
● Transcricións con gran precisión e integridade
● Altamente compatible con varias especies
● Gran capacidade de secuenciación con 4 plataformas de secuenciación PacBio Sequel II equipadas
● Moita experiencia con máis de 700 proxectos de secuenciación de ARN baseados en Pacbio
● Entrega de resultados baseada en BMKCloud: minería de datos personalizada dispoñible na plataforma.
● Servizos posvenda válidos durante 3 meses tras a finalización do proxecto
Especificacións do servizo
Plataforma: PacBio Sequel II
Biblioteca de secuenciación: biblioteca de ARNm enriquecida con Poly A
Rendemento de datos recomendado: 20 Gb/mostra (dependendo da especie)
FLNC(%): ≥75%
*FLNC: transciptos non quiméricos de lonxitude completa
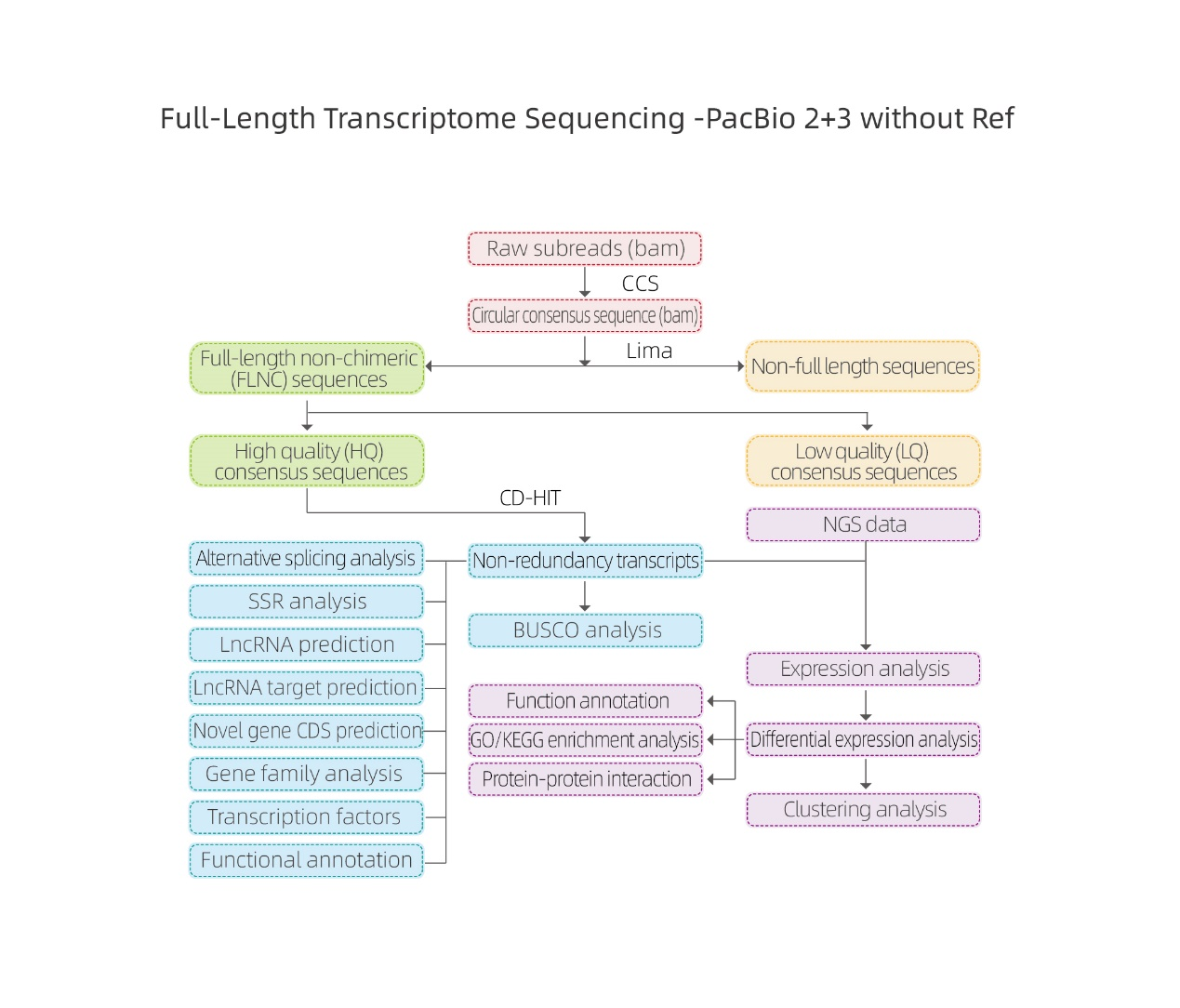
Análise bioinformática
● Tratamento de datos en bruto
● Identificación da transcrición
● Estrutura secuencial
● Cuantificación de expresións
● Anotación de funcións
Requisitos de mostra e entrega
Requisitos de mostra:
Nucleótidos:
| Conc. (ng/μl) | Cantidade (μg) | Pureza | Integridade |
| ≥ 120 | ≥ 0,6 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Contaminación limitada ou nula de proteínas ou ADN no xel. | Para plantas: RIN≥7,5; Para animais: RIN≥8,0; 5,0≥ 28S/18S≥1,0; elevación de referencia limitada ou nula |
Tecido: Peso (seco):≥ 1 g
*Para tecidos inferiores a 5 mg, recomendamos enviar mostras de tecido conxelada instantáneamente (en nitróxeno líquido).
Suspensión celular:Número de células = 3×106- 1×107
*Recomendamos enviar lisado celular conxelado.No caso de que esa cela conte menos de 5×105, recoméndase conxelar flash en nitróxeno líquido, o que é preferible para a micro extracción.
Mostras de sangue:Volume ≥1 ml
Microorganismo:Masa ≥ 1 g
Entrega de mostra recomendada
Envase:
Tubo de centrífuga de 2 ml (non se recomenda papel de aluminio)
Etiquetaxe da mostra: grupo+réplica, por exemplo, A1, A2, A3;B1, B2, B3....
Envío:
1. Xeo seco: as mostras deben ser embaladas en bolsas e enterradas en xeo seco.
2. Tubos RNAstable: as mostras de ARN pódense secar nun tubo de estabilización de ARN (por exemplo, RNAstable®) e enviarse a temperatura ambiente.
Fluxo de traballo do servizo

Deseño de experimentos

Entrega da mostra

extracción de ARN

Construción da biblioteca

Secuenciación

Análise de datos

Servizos posvenda
1.Distribución de lonxitude FLNC
A lonxitude da lectura non quimérica de lonxitude completa (FLNC) indica a lonxitude do ADNc na construción da biblioteca.A distribución de lonxitudes FLNC é un indicador crucial para avaliar a calidade da construción da biblioteca.
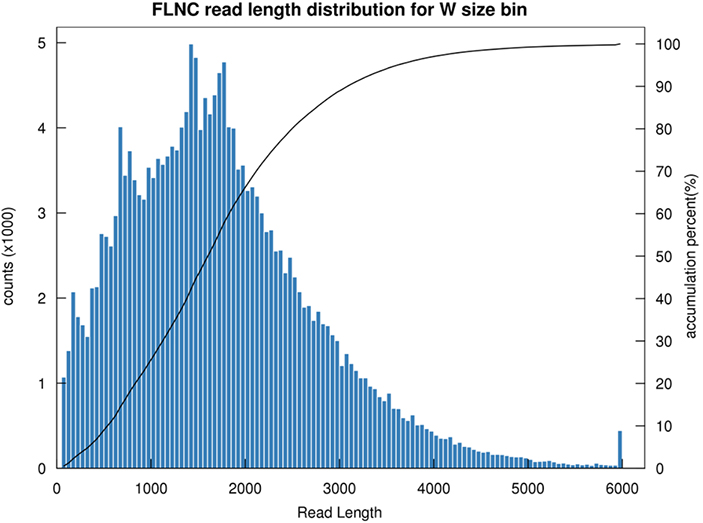
Distribución de lonxitude de lectura FLNC
2. Distribución completa da lonxitude da rexión ORF
Usamos TransDecoder para predicir rexións codificantes de proteínas e secuencias de aminoácidos correspondentes para xerar conxuntos uníxenos, que contén información completa de transcrición non redundante en todas as mostras.
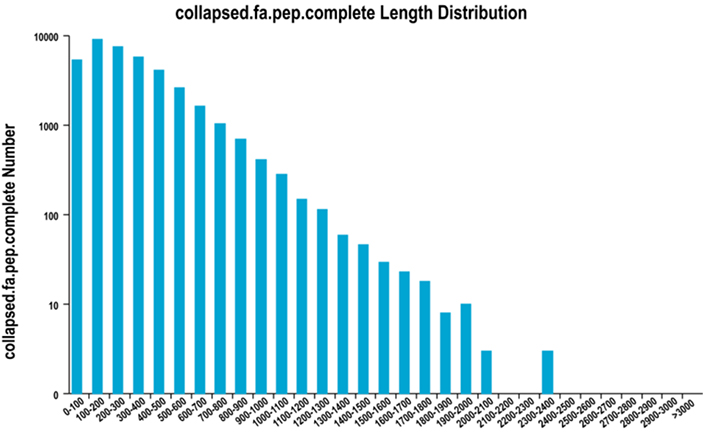
Distribución completa da lonxitude da rexión ORF
3.Análise do enriquecemento da vía KEGG
As transcricións expresadas de forma diferencial (DET) pódense identificar aliñando os datos de secuenciación de ARN baseados en NGS en conxuntos de transcricións de lonxitude completa xerados polos datos de secuenciación de PacBio.Estes DET pódense procesar máis adiante para varias análises funcionais, por exemplo, análise de enriquecemento da vía KEGG.
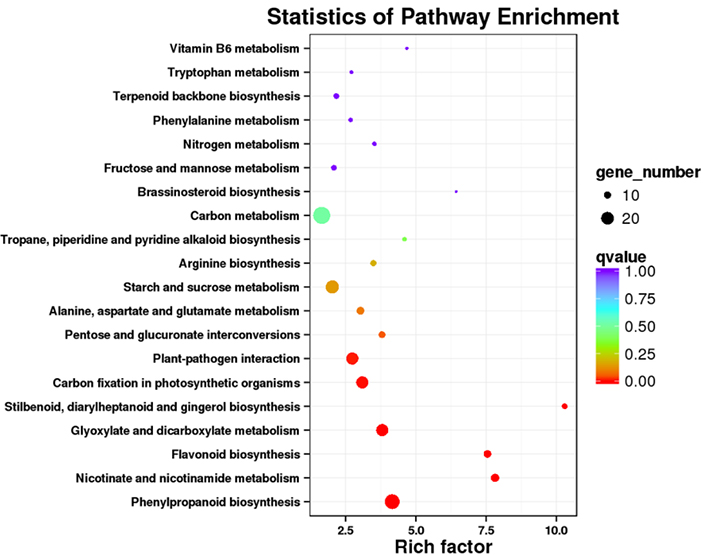
Enriquecemento da vía DET KEGG -Gráfica de puntos
Caso BMK
A dinámica de desenvolvemento do transcriptoma do tronco de Populus
Publicado: Revista de Biotecnoloxía Vexetal, 2019
Estratexia de secuenciación:
Recollida de mostras:rexións do tronco: ápice, primeiro entrenudo (IN1), segundo entrenudo (IN2), terceiro entrenudo (IN3), entrenudo (IN4) e entrenudo (IN5) de Nanlin895
Secuencia NGS:Agrupáronse ARN de 15 individuos como unha mostra biolóxica.Tres réplicas biolóxicas de cada punto foron procesadas para a secuencia NGS
Secuencia TGS:As rexións do tronco dividíronse en tres rexións, é dicir, ápice, IN1-IN3 e IN4-IN5.Cada rexión foi procesada para a secuenciación PacBio con catro tipos de bibliotecas: 0-1 kb, 1-2 kb, 2-3 kb e 3-10 kb.
Resultados clave
1. Identificáronse un total de 87150 transcricións completas, nas que se identificaron 2081 isoformas novas e 62058 isoformas empalmadas alternativas novas.
Identificáronse 2,1187 lncRNA e 356 xenes de fusión.
3.Do crecemento primario ao crecemento secundario, identificáronse 15838 transcricións expresadas de forma diferencial de 995 xenes expresados de forma diferencial.En todos os DEG, 1216 foron factores de transcrición, a maioría dos cales aínda non foron informados.
A análise de enriquecemento 4.GO revelou a importancia da división celular e do proceso de oxidación-redución no crecemento primario e secundario.
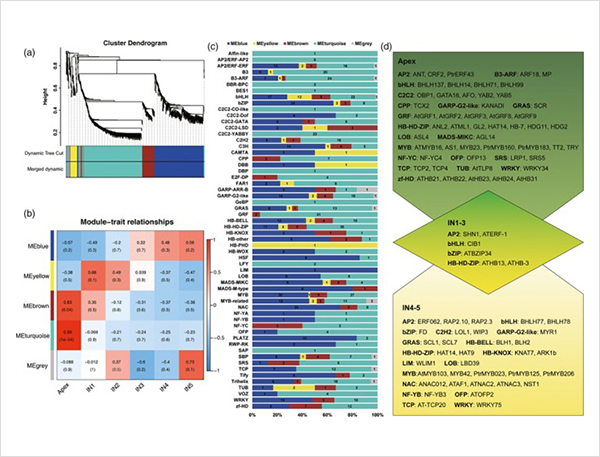
Eventos de empalme alternativos e diferentes isoformas
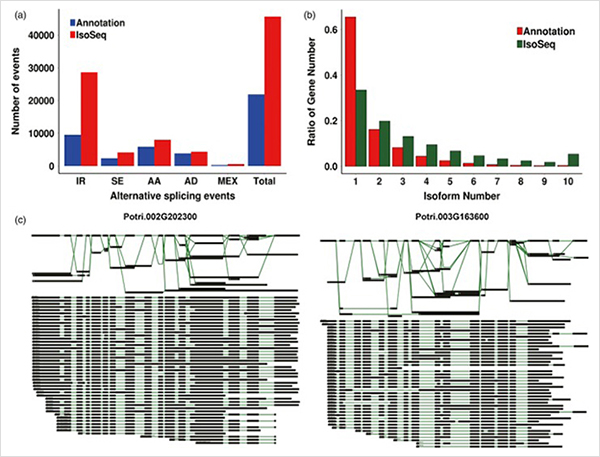
Análise WGCNA sobre factores de transcrición
Referencia
Chao Q, Gao ZF, Zhang D, et al.A dinámica de desenvolvemento do transcriptoma do tronco de Populus.Plant Biotechnol J. 2019;17(1):206-219.doi:10.1111/pbi.12958





