

Análise de segregación masiva






Vantaxes do servizo
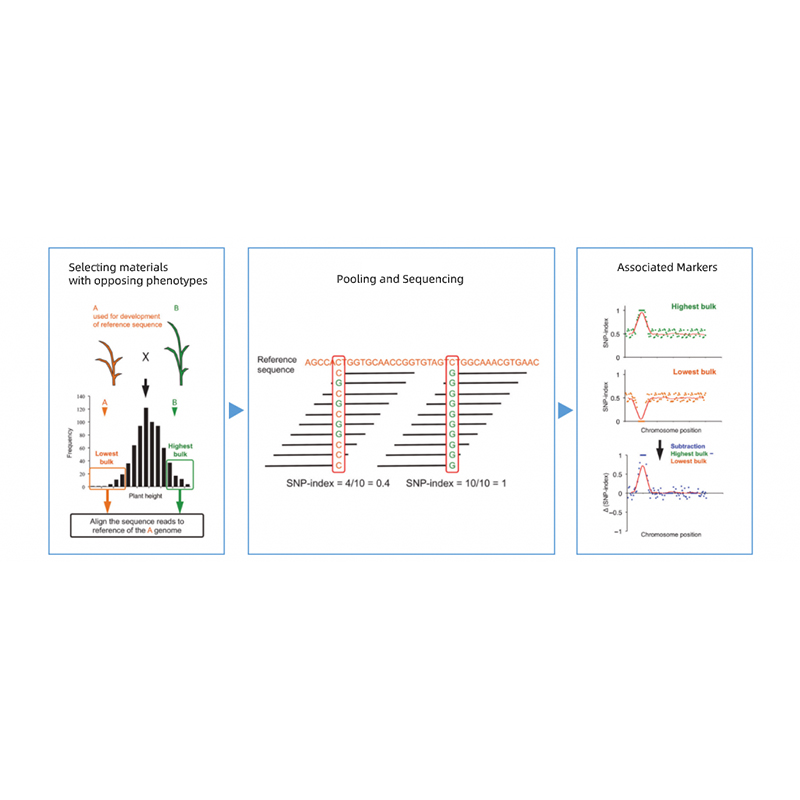
Takagi et al., The plant journal, 2013
● Localización precisa: mestura de masas con 30+30 a 200+200 individuos para minimizar o ruído de fondo;predicción da rexión candidata baseada en mutantes non sinónimos.
● Análise exhaustiva: anotación detallada da función do xene candidato, incluíndo NR, SwissProt, GO, KEGG, COG, KOG, etc.
● Tempo de resposta máis rápido: localización rápida dos xenes en 45 días hábiles.
● Ampla experiencia: BMK contribuíu na localización de miles de trazos, abranguendo especies diversas como cultivos, produtos acuáticos, bosques, flores, froitos, etc.
Especificacións do servizo
Poboación:
Proxenie segregadora de pais con fenotipos opostos.
por exemplo, proxenie F2, retrocruzamento (BC), liña endogámica recombinante (RIL)
Piscina de mestura
Para trazos cualitativos: 30 a 50 individuos (mínimo 20)/masa
Para tratos cuantitativos: entre o 5% e o 10% dos individuos con fenotipos extremos en toda a poboación (mínimo 30+30).
Profundidade de secuenciación recomendada
Polo menos 20X/pai e 1X/individuo descendente (por exemplo, para o grupo de mestura de descendencia de 30+30 individuos, a profundidade de secuenciación será de 30X por masa)
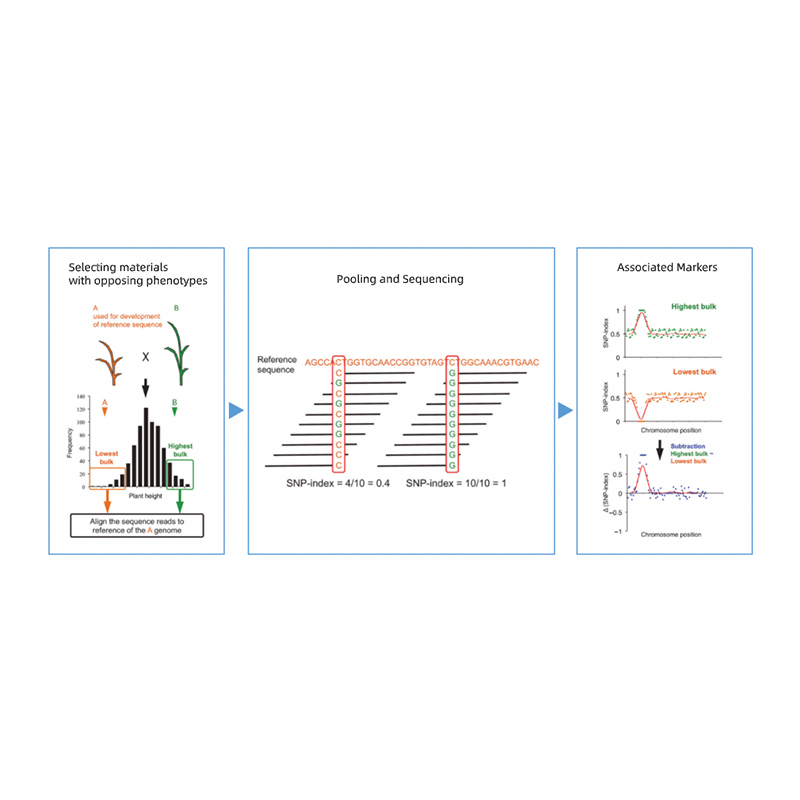
Análise bioinformática
● Resecuenciación do xenoma completo
● Tratamento de datos
● Chamada SNP/Indel
● Proba de rexións candidatas
● Anotación da función do xene candidato
Requisitos de mostra e entrega
Requisitos de mostra:
Nucleótidos:
| mostra de ADNg | Mostra de tecido |
| Concentración: ≥30 ng/μl | Plantas: 1-2 g |
| Cantidade: ≥2 μg (Volumen ≥15 μl) | Animais: 0,5-1 g |
| Pureza: OD260/280= 1,6-2,5 | Sangue enteiro: 1,5 ml |
Fluxo de traballo do servizo

Deseño de experimentos

Entrega da mostra

extracción de ARN

Construción da biblioteca

Secuenciación

Análise de datos

Servizos posvenda
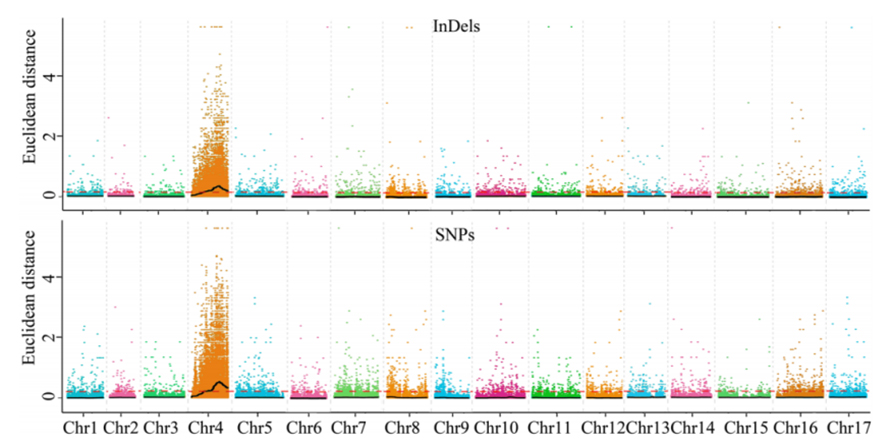
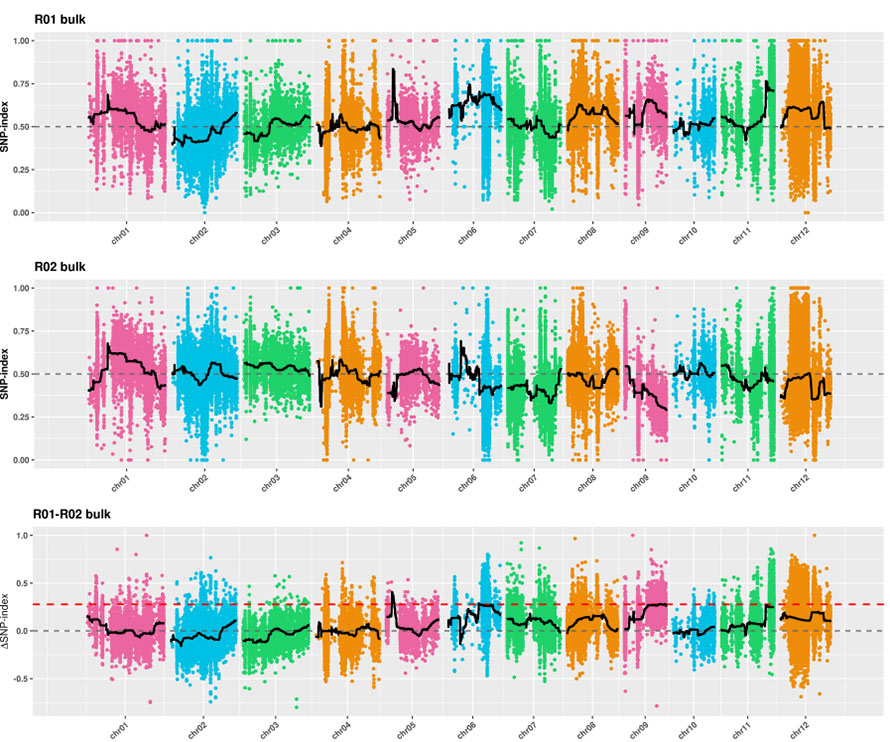
1.Análise da asociación baseada na Distancia Euclidiana (DE) para identificar a rexión candidata.Na seguinte figura
Eixe X: número de cromosomas;Cada punto representa un valor ED dun SNP.A liña negra corresponde ao valor ED axustado.Un valor de ED máis alto indica unha asociación máis significativa entre o sitio e o fenotipo.A liña trazos vermella representa o limiar de asociación significativa.
2.A análise da asociación baseada sen índice SNP
Eixe X: número de cromosomas;Cada punto representa o valor do índice SNP.A liña negra representa o valor do índice SNP axustado.Canto maior sexa o valor, máis significativa será a asociación.
Caso BMK
O locus de características cuantitativas de efecto principal Fnl7.1 codifica unha proteína abundante da embrioxénese tardía asociada á lonxitude do pescozo do froito no pepino.
Publicado: Revista de Biotecnoloxía Vexetal, 2020
Estratexia de secuenciación:
Pais (Jin5-508, YN): resecuenciación do xenoma completo para 34× e 20×.
Grupos de ADN (50 de pescozo longo e 50 de pescozo curto): resecuenciación para 61× e 52×
Resultados clave
Neste estudo, a poboación segregadora (F2 e F2:3) xerouse ao cruzar a liña de pepino de pescozo longo Jin5-508 e YN de pescozo curto.Dous pools de ADN foron construídos por 50 individuos de pescozo extremo extremo e 50 individuos de pescozo extremo extremo curto.O QTL de efecto maior identificouse en Chr07 mediante a análise de BSA e a cartografía QTL tradicional.A rexión candidata reduciuse aínda máis por cartografía fina, cuantificación da expresión xénica e experimentos transxénicos, que revelaron o xene clave para controlar a lonxitude do pescozo, CsFnl7.1.Ademais, descubriuse que o polimorfismo na rexión promotora de CsFnl7.1 estaba asociado coa expresión correspondente.Outras análises filoxenéticas suxiren que o locus Fnl7.1 é moi probable que se orixiñe da India.
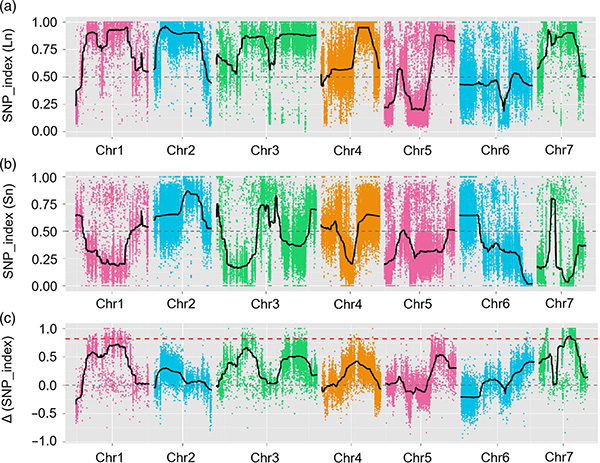 Mapeo QTL na análise BSA para identificar a rexión candidata asociada á lonxitude do pescozo do pepino | 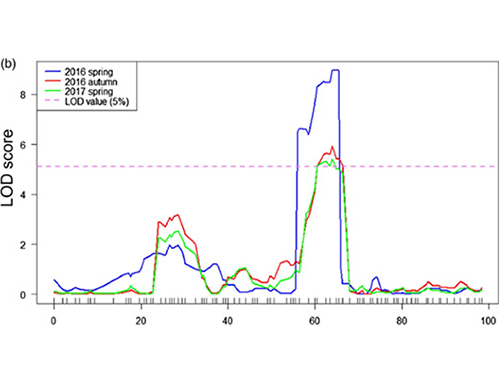 Perfís LOD de QTL de lonxitude do pescozo de pepino identificados en Chr07 |
Xu, X., et al."O locus do trazo cuantitativo de maior efecto Fnl7.1 codifica unha proteína abundante da embrioxénese tardía asociada á lonxitude do pescozo da froita no pepino".Revista de Biotecnoloxía Vexetal 18.7 (2020).





