

TRANSCRIPTOMIQUE
nature
COMMUNICATION
La caractérisation complète de la transcription de la mutation SF3B1 dans la leucémie lymphoïde chronique révèle une régulation négative des introns retenus
Transcriptions complètes|Séquençage des nanopores |Analyse isoforme alternative
Arrière-plan
SIl a été largement rapporté que des mutations omatiques du facteur d'épissage SF3B1 sont associées à divers cancers, notamment la leucémie lymphoïde chronique (LLC), le mélanome uvéal, le cancer du sein, etc. De plus, des études transcriptomiques à lecture courte ont révélé des modèles d'épissage aberrants induits par les mutations SF3B1.Cependant, les études sur ces modèles d'épissage alternatifs ont longtemps été limitées au niveau des événements et au manque de connaissances au niveau des isoformes en raison de la limitation des transcriptions assemblées à lecture courte.Ici, une plate-forme de séquençage de nanopores a été introduite pour générer des transcriptions complètes, ce qui a permis d'enquêter sur les isoformes AS.
Conception expérimentale
Expériences
Regroupement:1. CLL-SF3B1 (WT) 2. CLL-SF3B1 (mutation K700E) ;3. Cellules B normales
Stratégie de séquençage :Séquençage de bibliothèque MinION 2D, séquençage de bibliothèque PromethION 1D ;données à lecture courte à partir des mêmes échantillons
Plateforme de séquençage :ONT MinION ;ONT ProméthION ;
Analyse bioinformatique
Résultats
UNAu total, 257 millions de lectures ont été générées à partir de 6 échantillons de LLC et de 3 cellules B.En moyenne, 30,5 % de ces lectures ont été identifiées comme des transcriptions complètes.
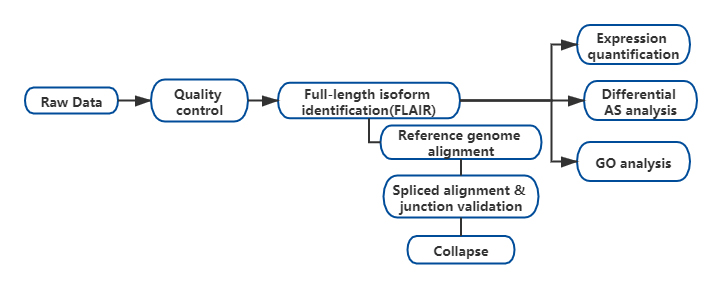
FL'analyse alternative des isoformes complètes de l'ARN (FLAIR) a été développée pour générer un ensemble d'isoformes de haute confiance.FLAIR peut se résumer comme suit :
Nanopore lit l'alignement : identifie la structure générale de la transcription en fonction du génome de référence ;
SCorrection des jonctions plice : corriger les erreurs de séquence (rouge) avec le site d'épissage provenant d'introns annotés, d'introns provenant de données à lecture courte ou des deux ;
Collapse : résumez les isoformes représentatives basées sur les chaînes de jonctions d'épissure (ensemble de premier passage).Sélectionnez une isofrom de confiance élevée en fonction du nombre de lectures prises en charge (seuil : 3).
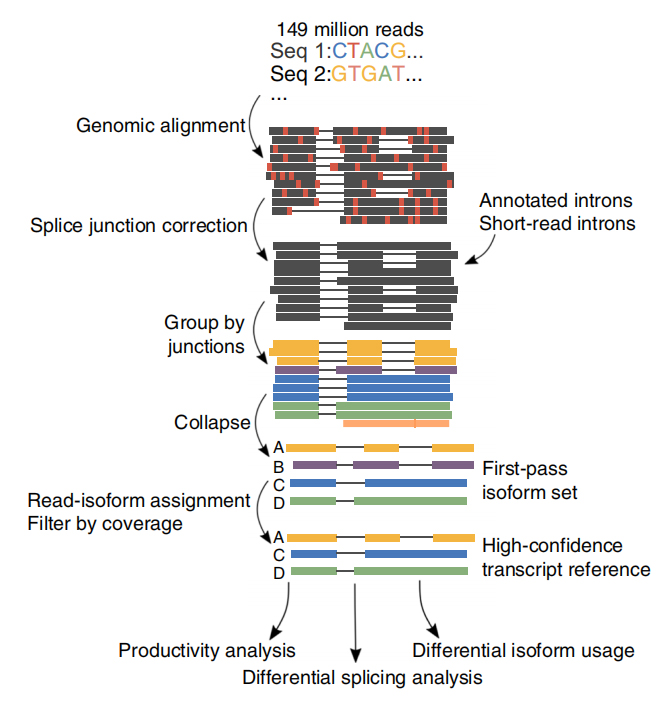
Figure 1. Analyse FLAIR pour identifier les isoformes complètes associées à la mutation SF3B1 dans la LLC
FLAIR a identifié 326 699 isoformes épissées avec un niveau de confiance élevé, dont 90 % sont de nouvelles isoformes.La plupart de ces isoformes non annotées se sont révélées être de nouvelles combinaisons de jonctions d'épissage connues (142 971), tandis que les autres nouvelles isoformes contenaient soit un intron retenu (21 700), soit un nouvel exon (3 594).
LLes séquences lues en continu permettent l'identification des sites d'épissage modifiés par SF3B1-K700E mutants au niveau de l'isoforme.Il a été constaté que 35 3'SS alternatifs et 10 5'SS alternatifs étaient épissés de manière significativement différentielle entre SF3B1-K700E et SF3B1-WT.33 des 35 altérations ont été nouvellement découvertes par des séquences à lecture longue.Dans les données Nanopore, la distribution de la distance entre les 3'SS modifiés par SF3B1-K700E et les pics des sites canoniques est d'environ -20 pb, ce qui diffère significativement d'une distribution témoin, similaire à ce qui a été rapporté dans les séquences de lecture courte de CLL.Les isoformes du gène ERGIC3 ont été analysées, où une nouvelle isoforme contenant le site d'épissage proximal s'est avérée plus abondante dans SF3B1-K700E.Les 3'SS proximaux et distaux étaient associés à des modèles AS distincts générant de multiples isoformes.
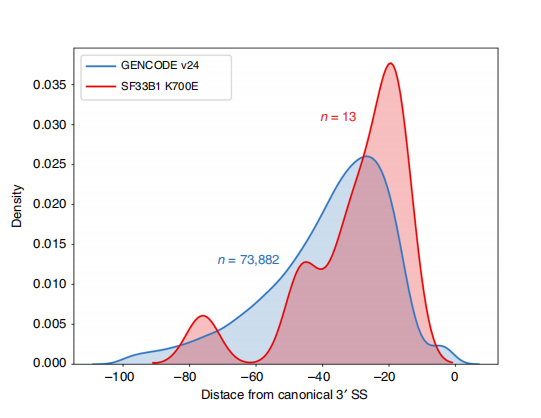
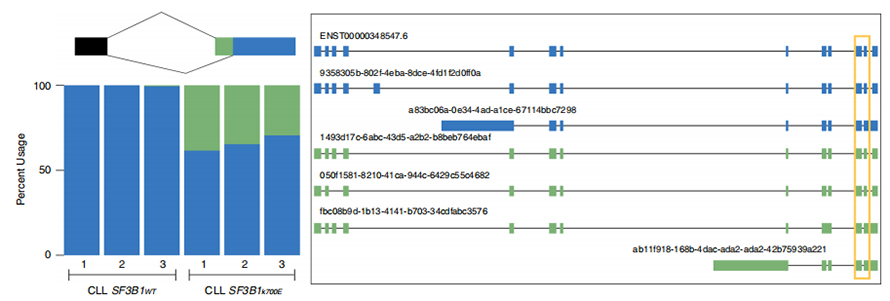
Figure 2. Modèles d'épissage 3' alternatifs identifiés avec les données de séquençage des nanopores
L'analyse de l'utilisation des événements IR a longtemps été limitée aux analyses basées sur des lectures courtes en raison de la confiance dans l'identification et la quantification des IR.L'expression des isoformes IR dans SF3B1-K700E et SF3B1-WT a été quantifiée sur la base de séquences de nanopores, révélant une régulation négative globale des isoformes IR dans SF3B1-K700E.
Figure 4. Intensité agricole et connectivité du réseau dans trois systèmes agricoles (A et B) ;Analyse forestière aléatoire (C) et relation entre l'intensité agricole et la colonisation par l'AMF (D)
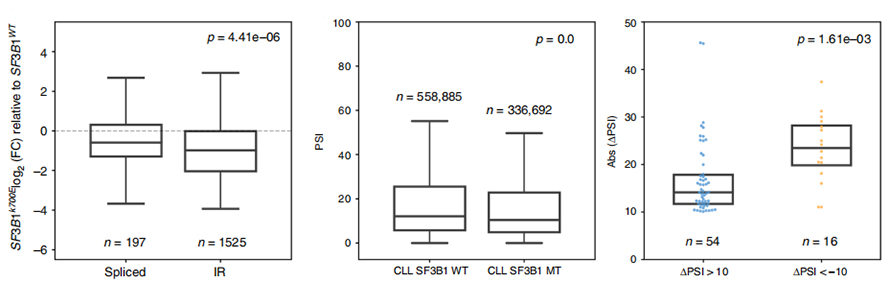
Figure 3. Les événements de rétention d'intron sont plus fortement régulés négativement dans la LLC SF3B1-K700E
Technologie
Séquençage à lecture longue de nanopores
NLe séquençage des anopores est une technologie de séquençage de signaux électriques en temps réel d’une seule molécule.
DL'ADN ou l'ARN double brin se liera aux protéines nanoporeuses intégrées dans le biofilm et se dérouleront sous la direction des protéines motrices.
DLes brins NA/ARN traversent la protéine du canal nanopore à un certain rythme sous l'action de la différence de tension.
Mles molécules génèrent différents signaux électriques selon la structure chimique.
Rla détection en temps réel des séquences est obtenue par appel de base.
Performance du séquençage complet du transcriptome
√ Saturation des données
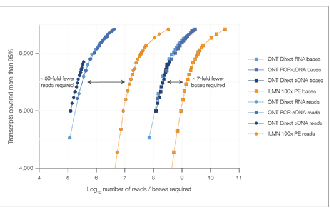
7 fois moins de lectures nécessaires pour atteindre une saturation de données comparable.
√ Identification de la structure de la transcription
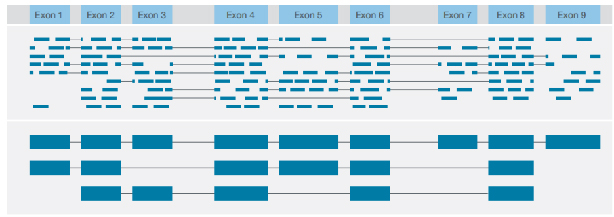
Identification de diverses variantes structurelles avec lecture consensuelle de chaque transcription
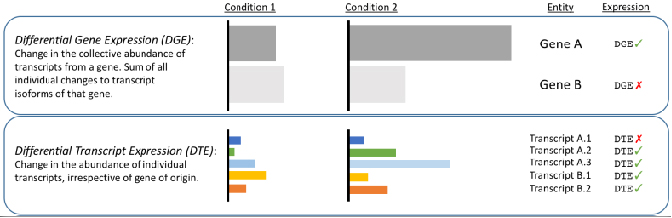
√ Analyse différentielle au niveau de la transcription - Révéler les changements masqués par les lectures courtes
Référence
Tang AD, Soulette CM, Baren MJV et al.La caractérisation complète de la transcription de la mutation SF3B1 dans la leucémie lymphoïde chronique révèle une régulation négative des introns retenus [J].Communication naturelle.
Technologie et points forts vise à partager les applications réussies les plus récentes de différentes technologies de séquençage à haut débit dans divers domaines de recherche ainsi que des idées brillantes en matière de conception expérimentale et d'exploration de données.
Heure de publication : 08 janvier 2022