

Assemblage du génome basé sur Hi-C







Avantages des services
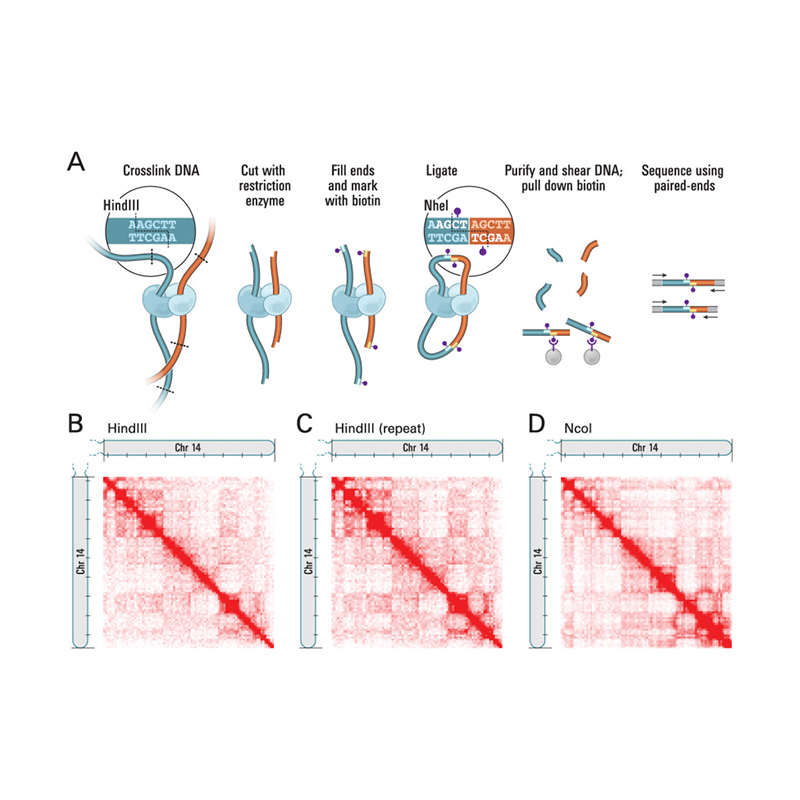
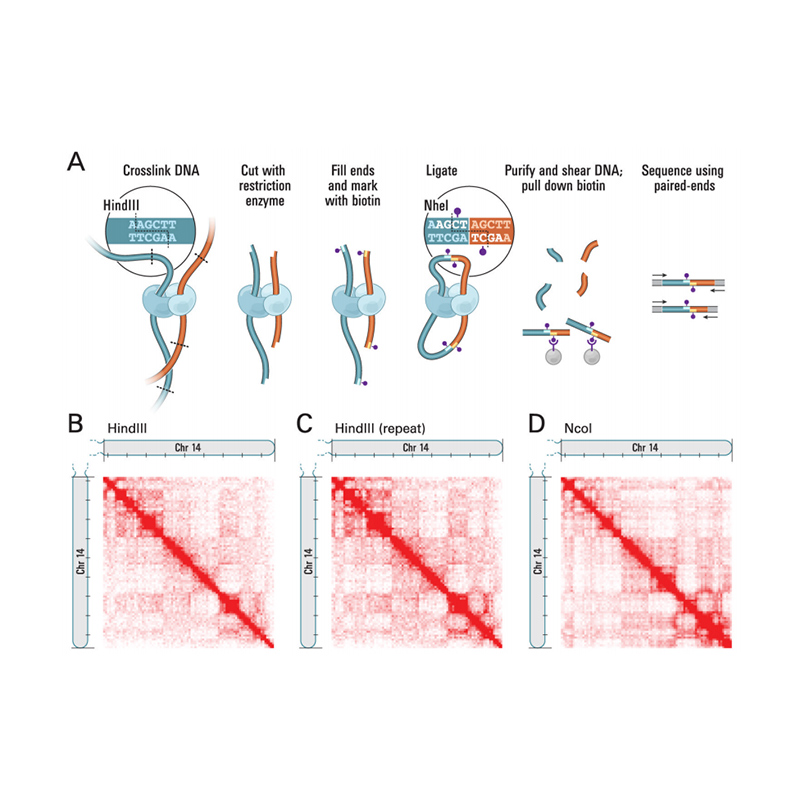
Présentation de Hi-C
(Lieberman-Aiden E et coll.,Science, 2009)
● Pas besoin de construire une population génétique pour l'ancrage contig ;
● Une densité de marqueurs plus élevée conduisant à un taux d'ancrage des contigs plus élevé, supérieur à 90 % ;
● Permet l'évaluation et les corrections sur les assemblages génomiques existants ;
● Délai d'exécution plus court avec une plus grande précision dans l'assemblage du génome ;
● Expérience abondante avec plus de 1 000 bibliothèques Hi-C construites pour plus de 500 espèces ;
● Plus de 100 cas réussis avec un facteur d'impact cumulatif publié de plus de 760 ;
● Assemblage du génome basé sur Hi-C pour le génome polyploïde, un taux d'ancrage de 100 % a été atteint dans le projet précédent ;
● Brevets internes et droits d'auteur sur les logiciels pour les expériences Hi-C et l'analyse des données ;
● Un logiciel de réglage des données visualisé auto-développé, permet le déplacement, l'inversion, la révocation et la refonte manuelle des blocs.
Spécifications des services
|
Type de bibliothèque
|
Plate-forme | Lire la longueur | Recommander une stratégie |
| Salut-C | Illumina NovaSeq | PE150 | ≥ 100X |
Analyses bioinformatiques
● Contrôle qualité des données brutes
● Contrôle qualité de la bibliothèque Hi-C
● Assemblage du génome basé sur Hi-C
● Évaluation post-assemblage
Exigences et livraison des échantillons
Exigences de l'échantillon :
| Animal | Champignon | Plantes
|
| Tissu congelé : 1 à 2 g par bibliothèque Cellules : 1x 10^7 cellules par bibliothèque | Tissu congelé : 1 g par bibliothèque | Tissu congelé : 1 à 2 g par bibliothèque
|
| *Nous recommandons fortement d'envoyer au moins 2 aliquotes (1 g chacune) pour l'expérience Hi-C. | ||
Livraison d’échantillon recommandée
Récipient : tube à centrifuger de 2 ml (le papier d'aluminium n'est pas recommandé)
Pour la plupart des échantillons, nous recommandons de ne pas conserver dans l'éthanol.
Étiquetage des échantillons : les échantillons doivent être clairement étiquetés et identiques au formulaire d’informations sur les échantillons soumis.
Expédition : Glace carbonique : Les échantillons doivent d’abord être emballés dans des sacs et enterrés dans de la glace carbonique.
Flux de travail des services

Conception d'expériences

Livraison d'échantillon

Extraction d'ADN

Construction d'une bibliothèque

Séquençage

L'analyse des données

Services après-vente
*Les résultats de démonstration présentés ici proviennent tous de génomes publiés avec Biomarker Technologies
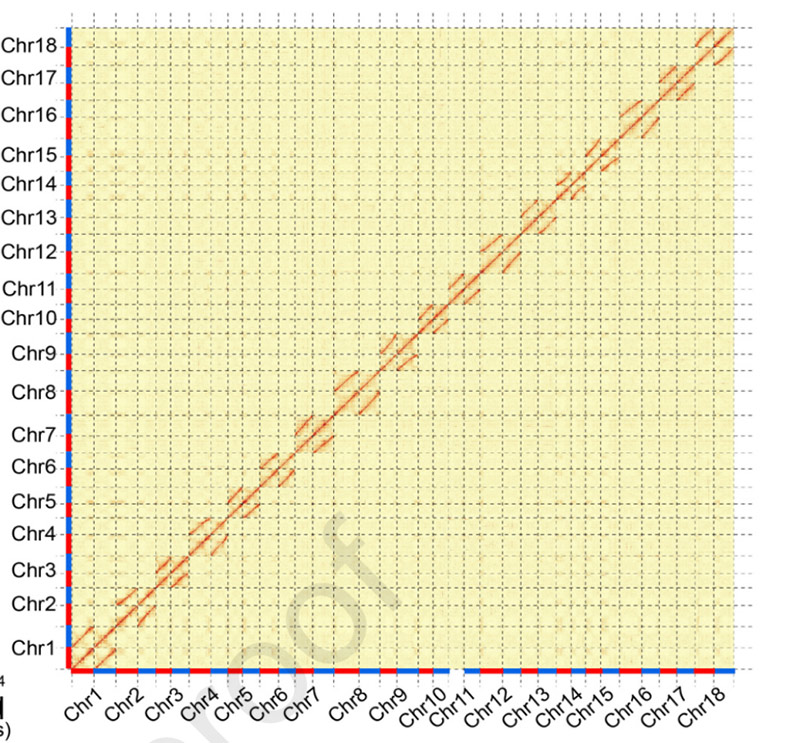
1. Carte thermique d'interaction Hi-C deCamptothèque acuminéegénome.Comme le montre la carte, l’intensité des interactions est négativement corrélée à la distance linéaire, ce qui indique un assemblage très précis au niveau des chromosomes.(Taux d'ancrage : 96,03%)
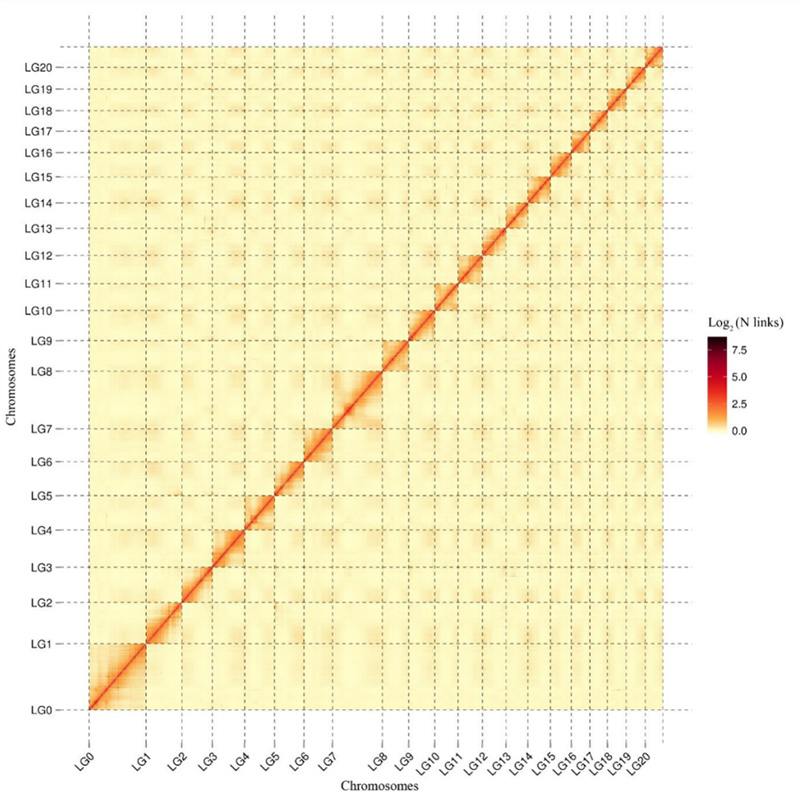
Kang M et coll.,Communications naturelles, 2021
2.Hi-C a facilité la validation des inversions entreGossypium hirsutumL.TM-1 A06 etG. arboreumChr06
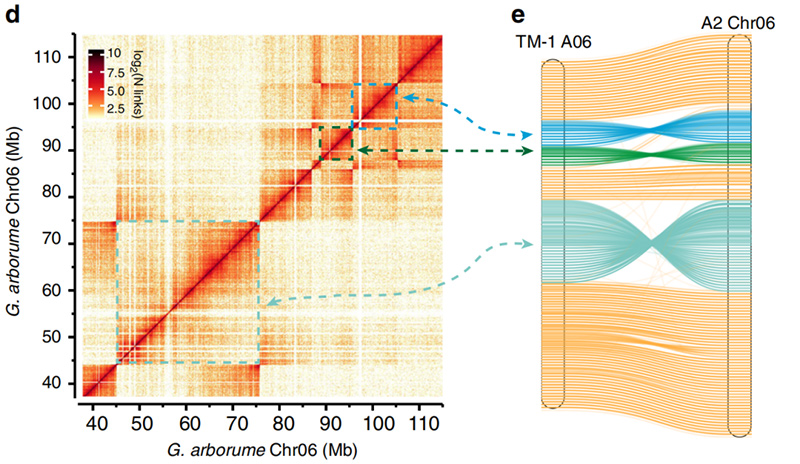
Yang Z et coll.,Communications nature, 2019
3.Assemblage et différenciation biallélique du génome du manioc SC205.La carte thermique Hi-C montre une division nette des chromosomes homologues.
Hu W et coll.,Plante moléculaire, 2021
4. Carte thermique Hi-C sur l'assemblage du génome de deux espèces de Ficus :F. microcarpa(taux d'ancrage : 99,3%) etF.hispida (taux d'ancrage : 99,7 %)
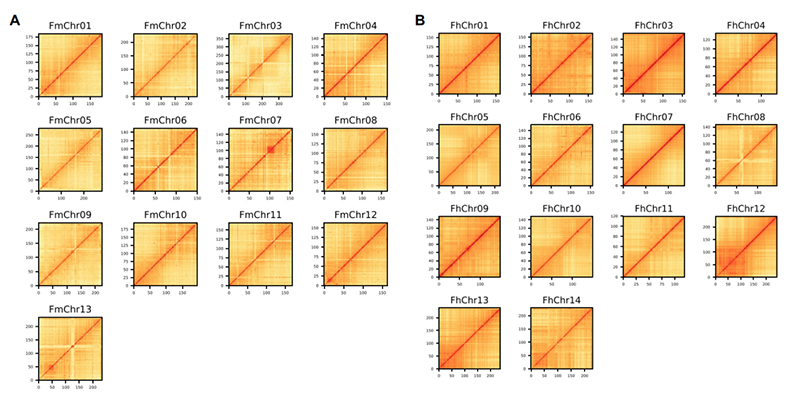
Zhang X et coll.,Cellule, 2020
Affaire BMK
Les génomes du banian et de la guêpe pollinisatrice fournissent un aperçu de la coévolution figue-guêpe
Publié : Cellule, 2020
Stratégie de séquençage :
F. microcarpa génome : env.84 X PacBio RSII (36,87 Go) + Hi-C (44 Go)
F. hispidagénome : env.97 X PacBio RSII (36,12 Go) + Hi-C (60 Go)
Eupristina verticilléegénome : env.170 X PacBio RSII (65 Go)
Résultats clés
1.Deux génomes de banian et un génome de guêpe pollinisatrice ont été construits à l'aide du séquençage PacBio, de Hi-C et d'une carte de liaison.
(1)F. microcarpagénome : Un assemblage de 426 Mo (97,7 % de la taille estimée du génome) a été établi avec un contig N50 de 908 Ko, un score BUSCO de 95,6 %.Au total, 423 séquences Mb ont été ancrées sur 13 chromosomes par Hi-C.L'annotation du génome a donné 29 416 gènes codant pour des protéines.
(2)F. Hispidagénome : Un assemblage de 360 Mb (97,3 % de la taille estimée du génome) a été obtenu avec un contig N50 de 492 Kb et un score BUSCO de 97,4 %.Un total de séquences de 359 Mb ont été ancrées sur 14 chromosomes par Hi-C et sont hautement identiques à la carte de liaison haute densité.
(3)Eupristina verticilléegénome : Un assemblage de 387 Mo (taille estimée du génome : 382 Mo) a été établi avec un contig N50 de 3,1 Mo et un score BUSCO de 97,7 %.
2. L'analyse génomique comparative a révélé un grand nombre de variations structurelles entre deuxFicusgénomes, qui ont fourni une ressource génétique inestimable pour les études d’évolution adaptative.Cette étude, pour la première fois, a fourni un aperçu de la coévolution figue-guêpe au niveau génomique.
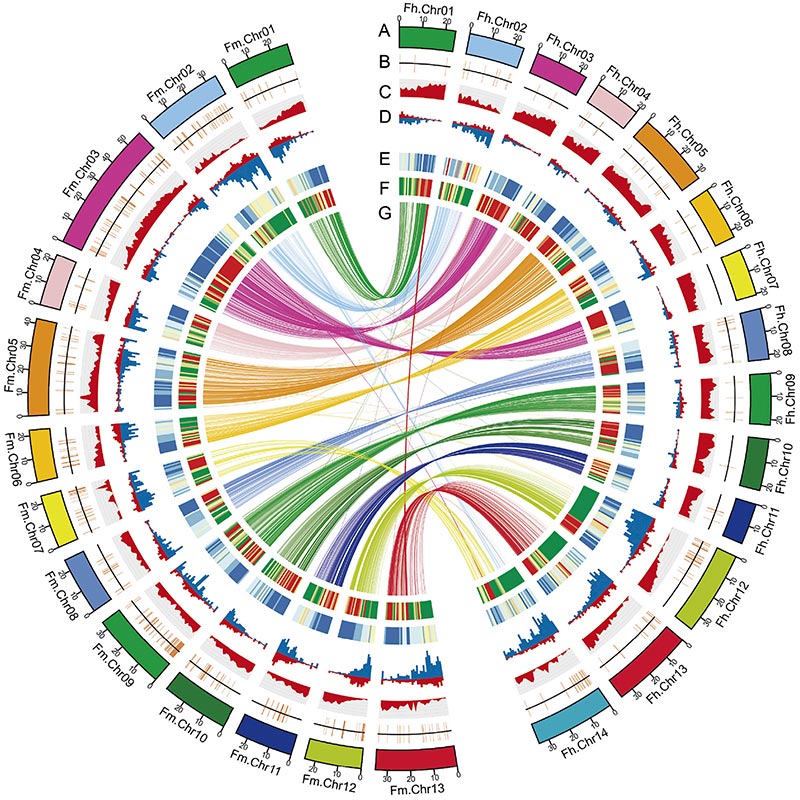 Diagramme Circos sur les caractéristiques génomiques de deuxFicusgénomes, y compris les chromosomes, les duplications segmentaires (SD), les transposons (LTR, TE, ADN TE), l'expression des gènes et la synténie | 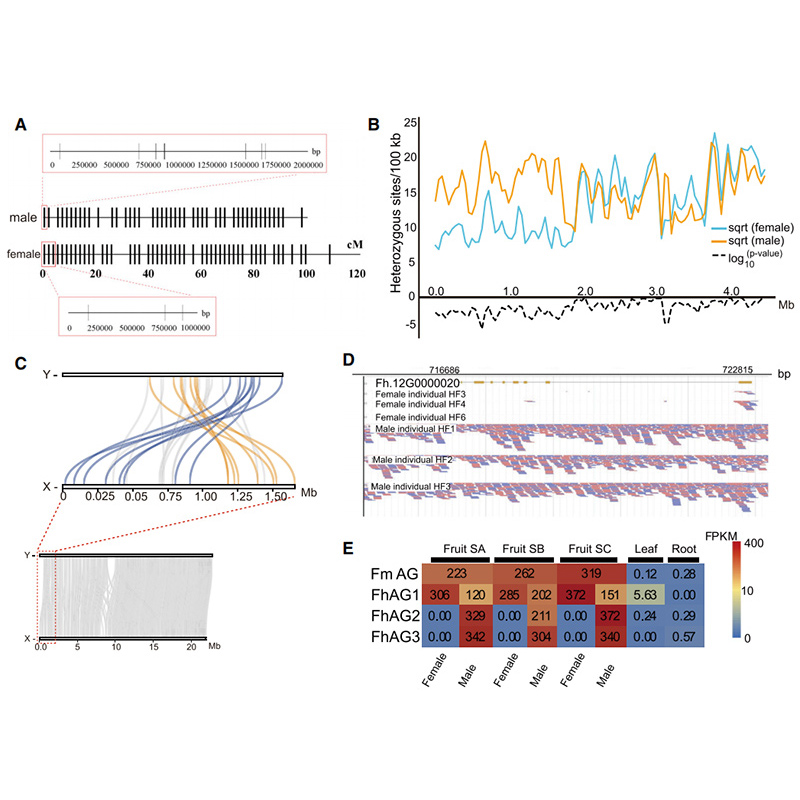 Identification du chromosome Y et du gène candidat à la détermination du sexe |
Zhang, X. et coll.«Les génomes du banian et de la guêpe pollinisatrice fournissent un aperçu de la coévolution figue-guêpe.»Cellule 183.4 (2020).





