

Génétique évolutive








Avantages des services
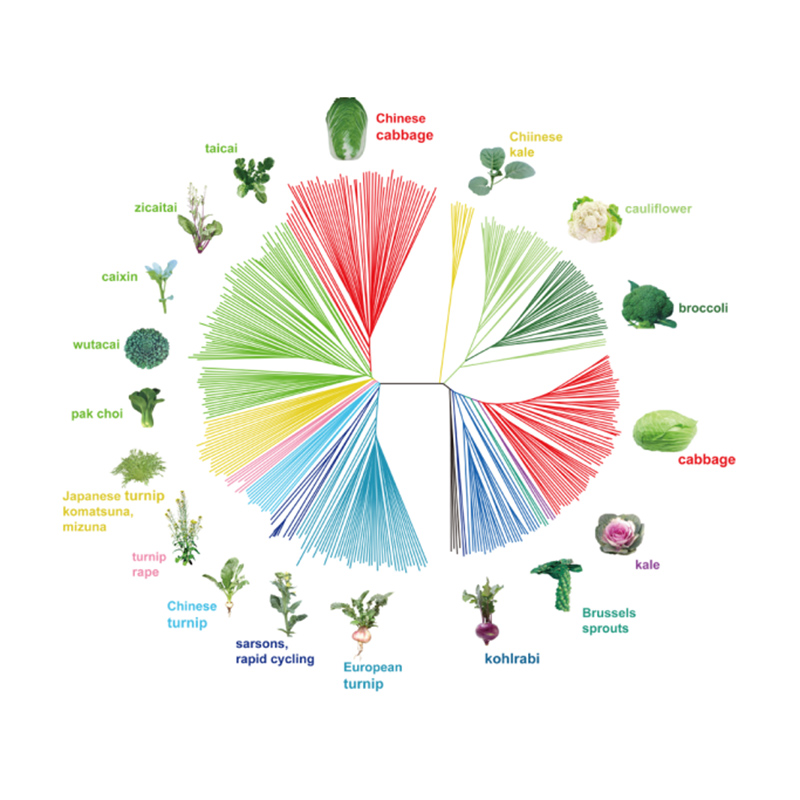
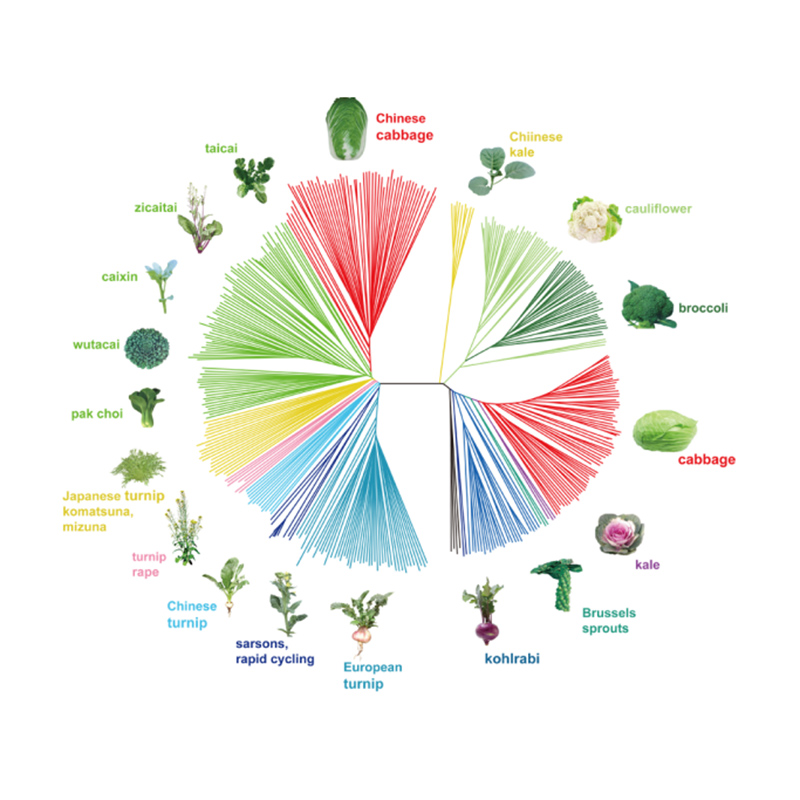
Takagi et coll.,Le journal des plantes, 2013
● Estimation du temps et de la vitesse de divergence des espèces en fonction des variations au niveau des nucléotides et des acides aminés
● Révélation d'une relation phylogénétique plus fiable entre les espèces avec une influence minimisée de l'évolution convergente et de l'évolution parallèle
● Construire des liens entre les changements génétiques et les phénotypes pour découvrir les gènes liés aux traits
● Estimation de la diversité génétique, qui reflète le potentiel évolutif des espèces
● Délai d'exécution plus rapide
● Vaste expérience : BMK a accumulé une expérience considérable dans des projets liés à la population et à l'évolution depuis plus de 12 ans, couvrant des centaines d'espèces, etc. et a contribué à plus de 80 projets de haut niveau publiés dans Nature Communications, Molecular Plants, Plant Biotechnology Journal, etc.
Spécifications des services
Matériaux:
Normalement, au moins trois sous-populations (par exemple sous-espèces ou souches) sont recommandées.Chaque sous-population ne doit pas contenir moins de 10 individus (Plantes >15, peut être réduit pour les espèces rares).
Stratégie de séquençage :
* WGS peut être utilisé pour les espèces dotées d'un génome de référence de haute qualité, tandis que SLAF-Seq est applicable aux espèces avec ou sans génome de référence, ou à un génome de référence de mauvaise qualité.
| Applicable à la taille du génome | WGS | Balises SLAF (×10 000) |
| ≤ 500 Mo | 10×/individu | WGS est plus recommandé |
| 500 Mo - 1 Go | 10 | |
| 1 Go - 2 Go | 20 | |
| ≥2 Go | 30 |
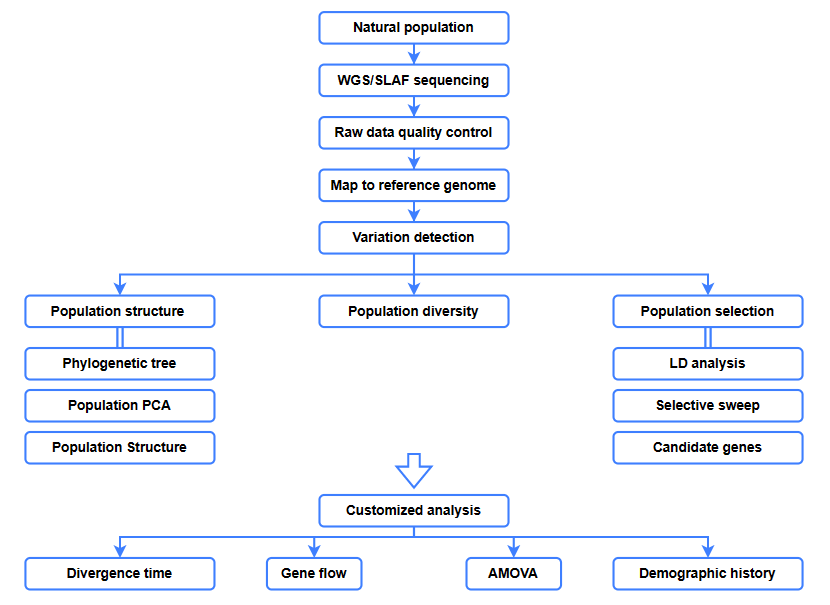
Analyses bioinformatiques
● Analyse évolutive
● Balayage sélectif
● Flux génétique
● Histoire démographique
● Temps de divergence
Exigences et livraison des échantillons
Exigences de l'échantillon :
| Espèces | Tissu | WGS-NGS | SLAF |
| Animal
| Tissu viscéral |
0,5 ~ 1g
|
0,5g
|
| Tissu musculaire | |||
| Sang de mammifère | 1,5 ml
| 1,5 ml
| |
| Sang de volaille/poisson | |||
| Usine
| Feuille fraîche | 1~2g | 0,5 ~ 1g |
| Pétale/Tige | |||
| Racine/graine | |||
| Cellules | Cellule cultivée |
| ADNg | Concentration | Montant (pouah) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1,6-2,5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Flux de travail des services

Conception d'expériences

Livraison d'échantillon

Construction d'une bibliothèque

Séquençage

L'analyse des données

Services après-vente
*Les résultats de démonstration présentés ici proviennent tous de génomes publiés avec BMKGENE
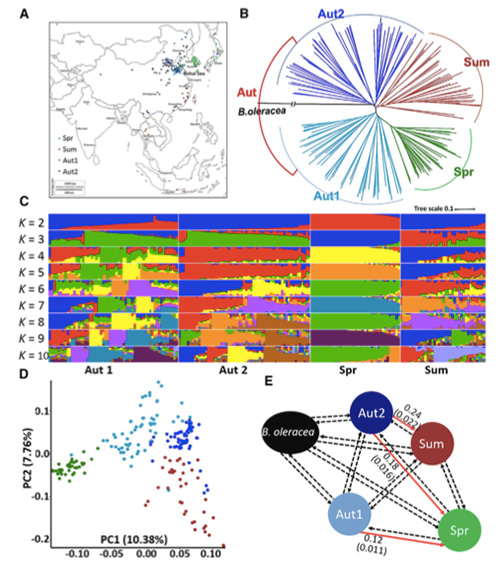
1. L'analyse de l'évolution comprend la construction d'un arbre phylogénétique, de la structure de la population et d'une PCA basée sur les variations génétiques.
L'arbre phylogénétique représente les relations taxonomiques et évolutives entre les espèces ayant un ancêtre commun.
PCA vise à visualiser la proximité entre les sous-populations.
La structure de la population montre la présence de sous-populations génétiquement distinctes en termes de fréquences alléliques.
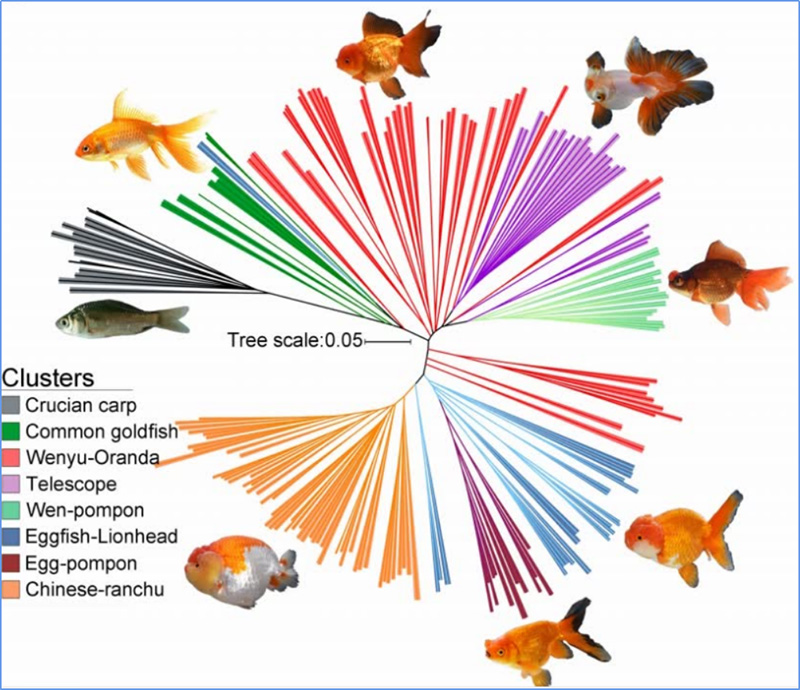
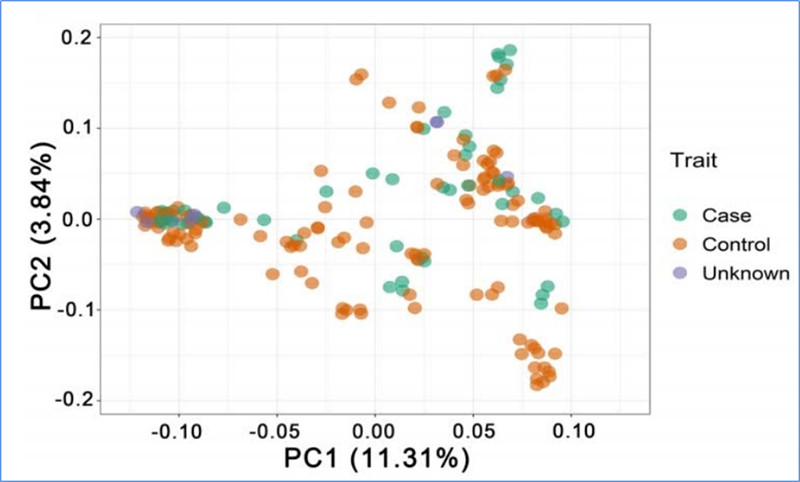
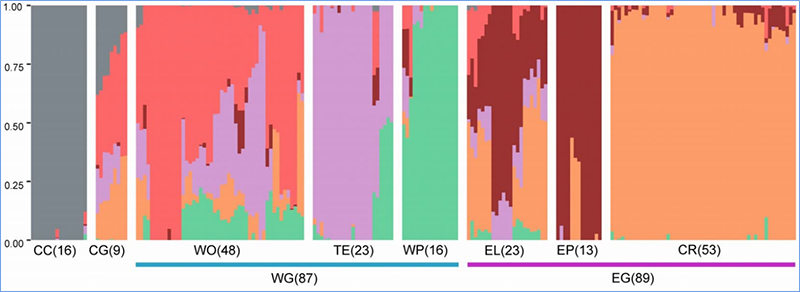
Chen, et.Al.,PNAS, 2020
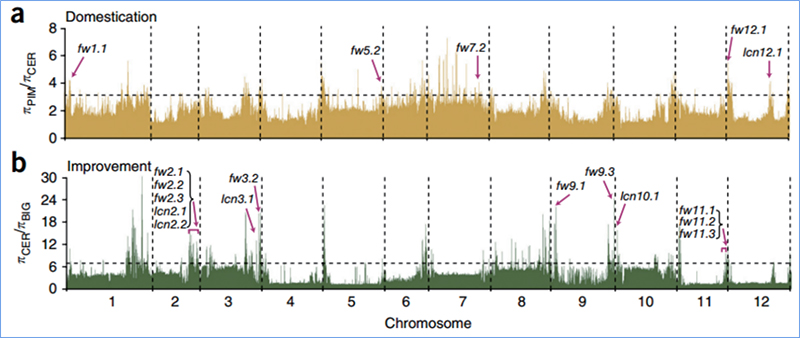
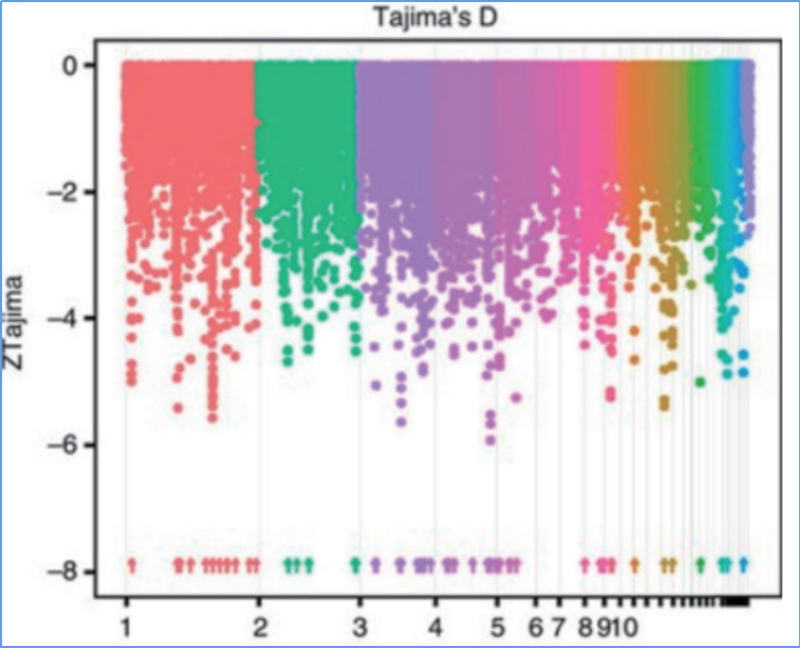
2. Balayage sélectif
Le balayage sélectif fait référence à un processus par lequel un site avantageux est sélectionné et les fréquences des sites neutres liés sont augmentées et celles des sites non liés sont diminuées, ce qui entraîne une réduction du niveau régional.
La détection à l'échelle du génome sur les régions de balayage sélectif est traitée en calculant l'indice génétique de la population (π, Fst, D de Tajima) de tous les SNP dans une fenêtre glissante (100 Ko) à une certaine étape (10 Ko).
Diversité nucléotidique(π)
Le D de Tajima
Indice de fixation (Fst)
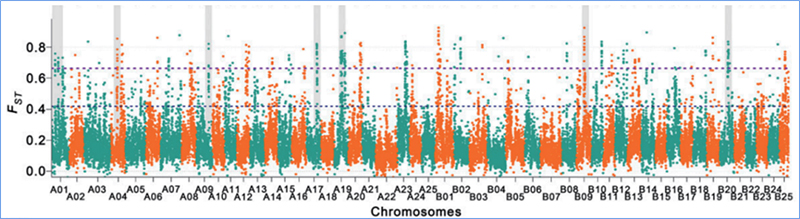
Wu, et.Al.,Plante moléculaire, 2018
3. Flux génétique
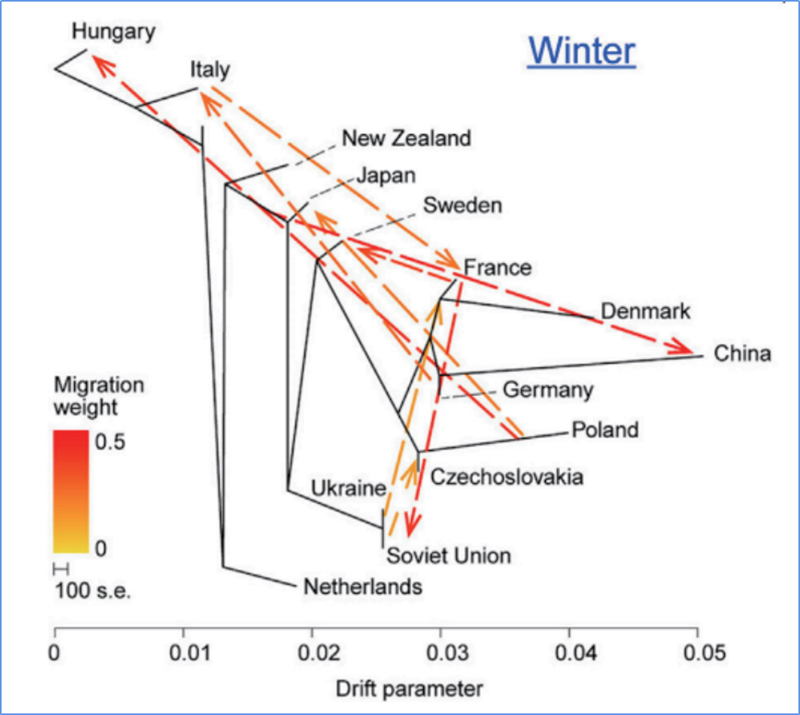
Wu, et.Al.,Plante moléculaire, 2018
4.Histoire démographique
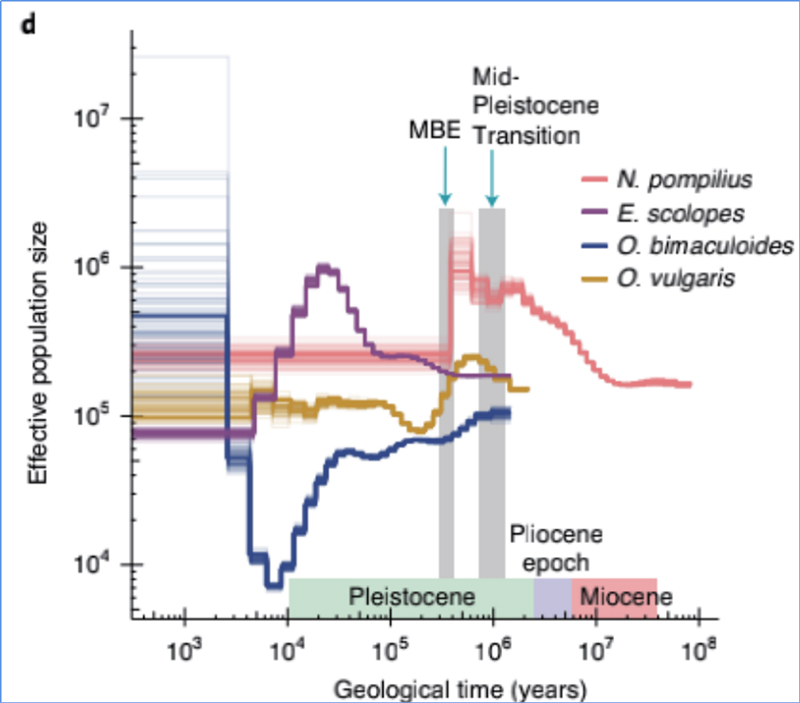
Zhang, et.Al.,Nature Ecologie&Evolution, 2021
5. Temps de divergence
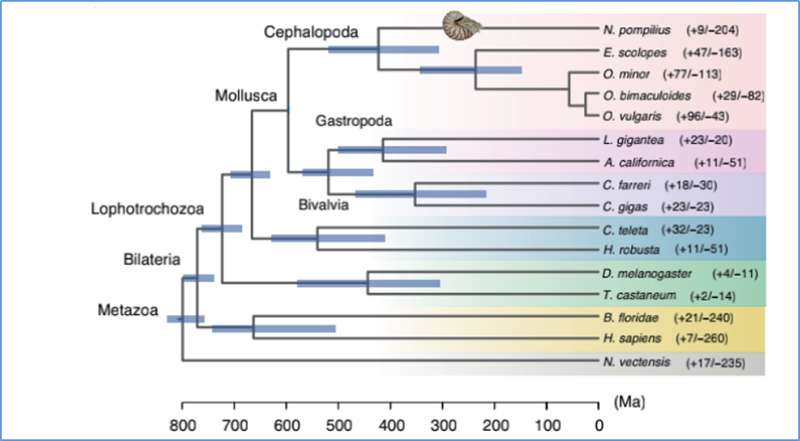
Zhang, et.Al.,Nature Ecologie&Evolution, 2021
Affaire BMK
Une carte de variation génomique fournit un aperçu de la base génétique de la sélection du chou chinois de printemps (Brassica rapa ssp. Pekinensis).
Publié : Plante moléculaire, 2018
Stratégie de séquençage :
Reséquençage : profondeur de séquençage : 10×
Résultats clés
Dans cette étude, 194 choux chinois ont été traités pour un re-séquençage avec une profondeur moyenne de 10×, ce qui a donné 1 208 499 SNP et 416 070 InDels.L'analyse phylogénétique de ces 194 lignées a montré que ces lignées peuvent être divisées en trois écotypes, printemps, été et automne.De plus, la structure de la population et l'analyse PCA ont indiqué que le chou chinois de printemps provenait d'un chou d'automne du Shandong, en Chine.Ceux-ci ont ensuite été introduits en Corée et au Japon, croisés avec des lignées locales et certaines variétés à montée tardive ont été réintroduites en Chine et sont finalement devenues du chou chinois de printemps.
Une analyse pangénomique des choux chinois de printemps et des choux d'automne sélectionnés a révélé 23 loci génomiques ayant subi une sélection forte, dont deux se chevauchaient avec une région de contrôle du temps de montaison basée sur la cartographie QTL.Ces deux régions contiennent des gènes clés qui régulent la floraison, BrVIN3.1 et BrFLC1.Il a en outre été confirmé que ces deux gènes étaient impliqués dans le temps de montaison par une étude du transcriptome et des expériences transgéniques.
 Analyse de la structure de la population de choux chinois | 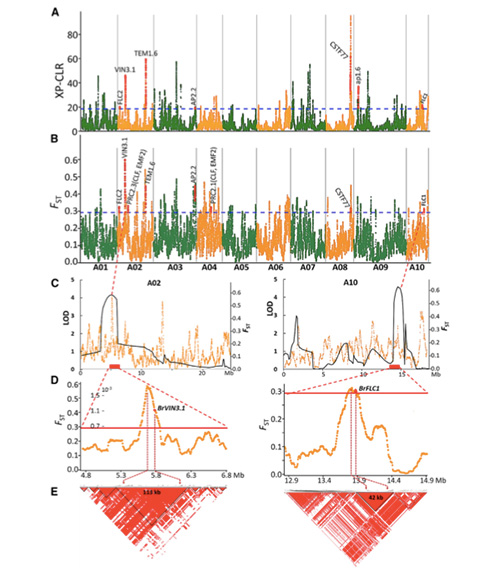 Informations génétiques sur la sélection du chou chinois |
Tongbing et coll.«Une carte de variation génomique fournit un aperçu de la base génétique de la sélection du chou chinois de printemps (Brassica rapa ssp.pekinensis).»Plantes moléculaires,11(2018):1360-1376.





