

Séquençage complet de l'ARNm -PacBio





Avantages des services
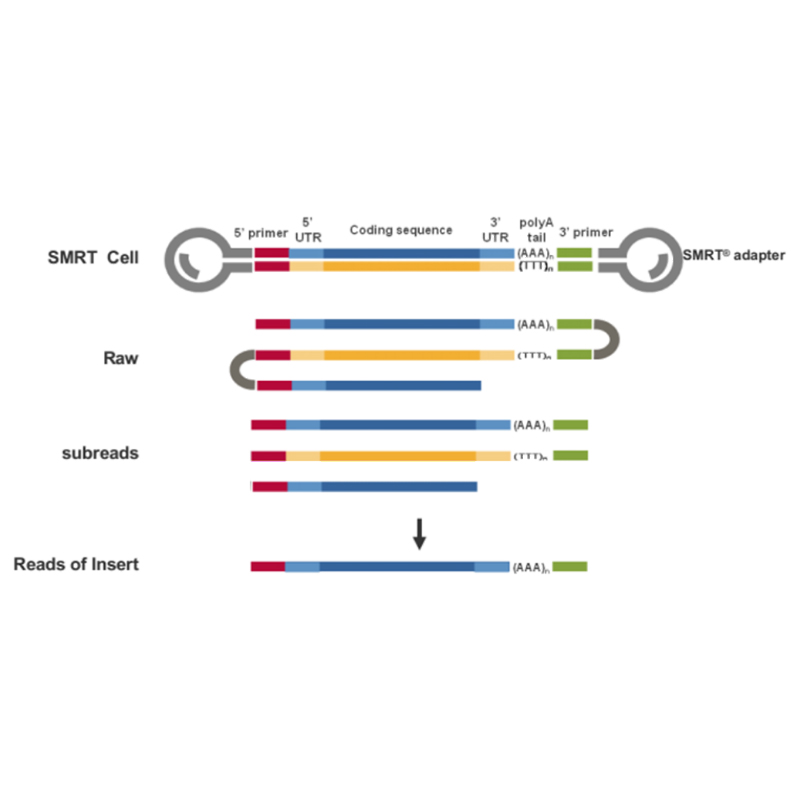
● Lecture directe de la molécule d'ADNc complète de l'extrémité 3' à l'extrémité 5'
● Résolution au niveau iso-forme dans la structure de séquence
● Transcriptions avec une grande précision et intégrité
● Hautement compatible avec les espèces vaieurs
● Grande capacité de séquençage avec 4 plateformes de séquençage PacBio Sequel II équipées
● Très expérimenté avec plus de 700 projets de séquençage d'ARN basés sur Pacbio
● Livraison de résultats basée sur BMKCloud : exploration de données personnalisée disponible sur la plateforme.
● Services après-vente valables 3 mois après la fin du projet
Spécifications des services
Plateforme : PacBio Sequel II
Bibliothèque de séquençage : bibliothèque d'ARNm enrichie en Poly A
Rendement de données recommandé : 20 Go/échantillon (selon l'espèce)
FLNC(%):≥75%
*FLNC : transcriptions complètes non chimériques
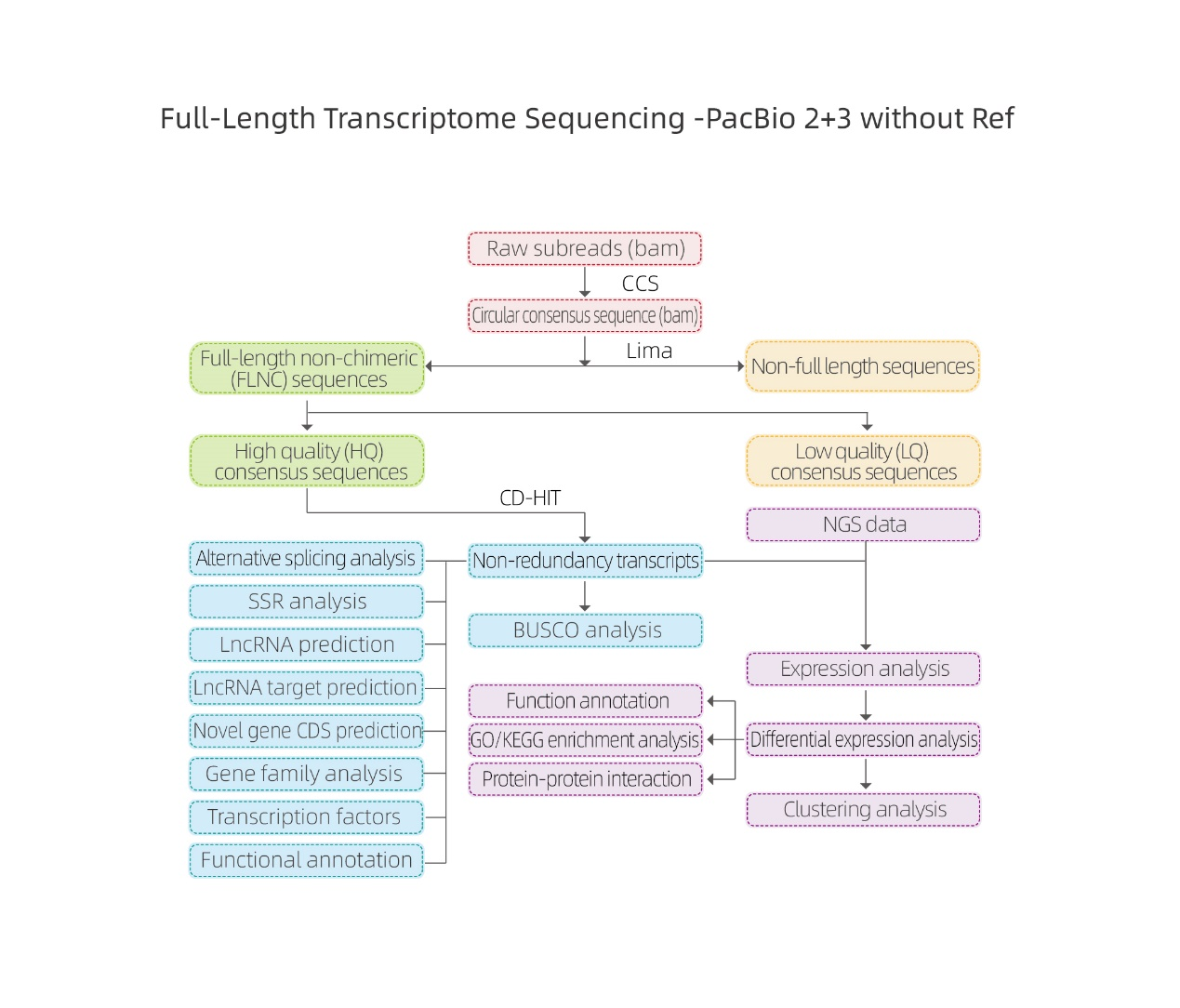
Analyses bioinformatiques
● Traitement des données brutes
● Identification du relevé de notes
● Structure de séquence
● Quantification des expressions
● Annotation de fonction
Exigences et livraison des échantillons
Exigences de l'échantillon :
Nucléotides :
| Conc. (ng/μl) | Quantité (μg) | Pureté | Intégrité |
| ≥120 | ≥ 0,6 | DO260/280=1,7-2,5 DO260/230=0,5-2,5 Contamination limitée ou inexistante en protéines ou en ADN indiquée sur le gel. | Pour les plantes : RIN≥7,5 ; Pour les animaux : RIN≥8,0 ; 5,0≥28S/18S≥1,0 ; élévation de la ligne de base limitée ou inexistante |
Tissu : Poids (sec) :≥1 g
*Pour les tissus inférieurs à 5 mg, nous recommandons d’envoyer un échantillon de tissu surgelé (dans l’azote liquide).
Suspension cellulaire :Nombre de cellules = 3×106- 1×107
*Nous recommandons d’expédier du lysat cellulaire congelé.Dans le cas où cette cellule compte moins de 5×105, une surgélation dans l'azote liquide est recommandée, ce qui est préférable pour la micro-extraction.
Échantillons de sang:Volume≥1 mL
Micro-organisme:Masse ≥ 1 g
Livraison d’échantillon recommandée
Récipient:
Tube à centrifuger de 2 ml (le papier d’aluminium n’est pas recommandé)
Étiquetage des échantillons : Groupe + répétition, par exemple A1, A2, A3 ;B1, B2, B3... ...
Expédition:
1. Glace sèche : Les échantillons doivent être emballés dans des sacs et enterrés dans de la neige carbonique.
2. Tubes RNAstable : les échantillons d'ARN peuvent être séchés dans un tube de stabilisation d'ARN (par exemple RNAstable®) et expédiés à température ambiante.
Flux de travail des services

Conception d'expériences

Livraison d'échantillon

Extraction d'ARN

Construction d'une bibliothèque

Séquençage

L'analyse des données

Services après-vente
1. Répartition des longueurs FLNC
La longueur de la lecture non chimérique complète (FLNC) indique la longueur de l'ADNc dans la construction de la bibliothèque.La distribution de longueur FLNC est un indicateur crucial dans l’évaluation de la qualité de la construction de la bibliothèque.
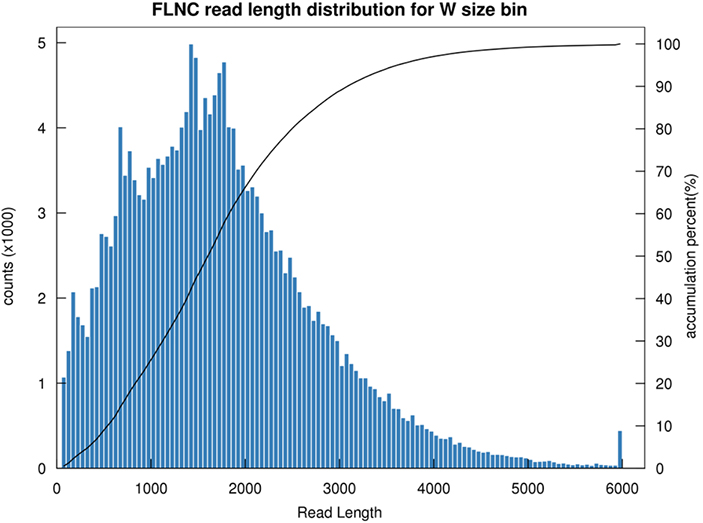
Distribution de longueur de lecture FLNC
2. Distribution complète de la longueur de la région ORF
Nous utilisons TransDecoder pour prédire les régions codantes pour les protéines et les séquences d'acides aminés correspondantes afin de générer des ensembles unigènes, qui contiennent des informations de transcription complètes et non redondantes dans tous les échantillons.
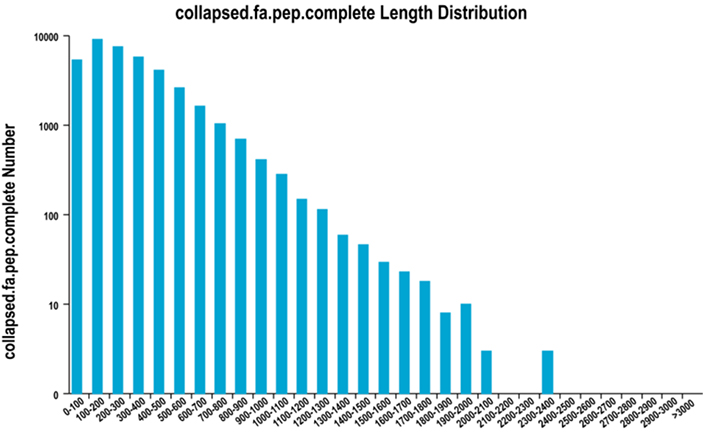
Distribution complète de la longueur de la région ORF
Analyse d'enrichissement de la voie 3.KEGG
Les transcriptions différentiellement exprimées (DET) peuvent être identifiées en alignant les données de séquençage d'ARN basées sur NGS sur les ensembles de transcriptions complètes générés par les données de séquençage PacBio.Ces DET peuvent être traités ultérieurement pour diverses analyses fonctionnelles, par exemple l'analyse d'enrichissement de la voie KEGG.
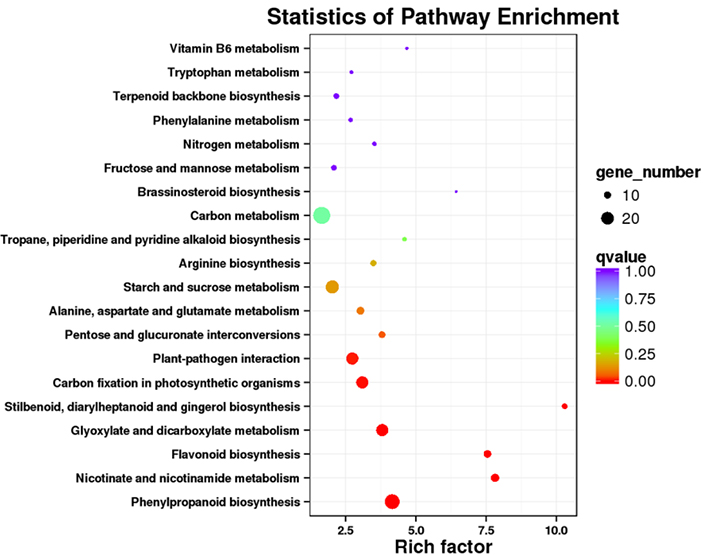
Enrichissement de la voie DET KEGG - Dot plot
Affaire BMK
La dynamique de développement du transcriptome de la tige de Populus
Publié : Journal de biotechnologie végétale, 2019
Stratégie de séquençage :
Collecte d'échantillons:régions de tige : sommet, premier entre-nœud (IN1), deuxième entre-nœud (IN2), troisième entre-nœud (IN3), entre-nœud (IN4) et entre-nœud (IN5) de Nanlin895
Séquence NGS :L'ARN de 15 individus a été regroupé en un seul échantillon biologique.Trois répliques biologiques de chaque point ont été traitées pour la séquence NGS
Séquence TGS :Les régions souches ont été divisées en trois régions, à savoir l'apex, IN1-IN3 et IN4-IN5.Chaque région a été traitée pour le séquençage PacBio avec quatre types de bibliothèques : 0-1 Ko, 1-2 Ko, 2-3 Ko et 3-10 Ko.
Résultats clés
1. Au total, 87 150 transcriptions complètes ont été identifiées, dans lesquelles 2 081 nouvelles isoformes et 62 058 nouvelles isoformes épissées alternatives ont été identifiées.
2,1187 lncRNA et 356 gènes de fusion ont été identifiés.
3. De la croissance primaire à la croissance secondaire, 15 838 transcrits différentiellement exprimés provenant de 995 gènes différentiellement exprimés ont été identifiés.Dans tous les DEG, 1 216 étaient des facteurs de transcription, dont la plupart n’ont pas encore été rapportés.
L'analyse d'enrichissement 4.GO a révélé l'importance de la division cellulaire et du processus d'oxydo-réduction dans la croissance primaire et secondaire.
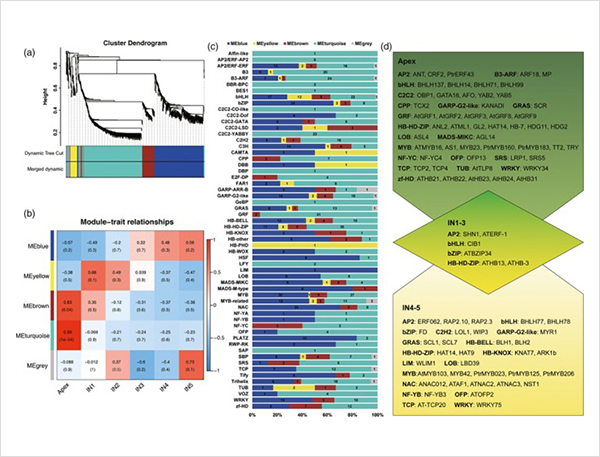
Événements d'épissage alternatifs et différentes isoformes
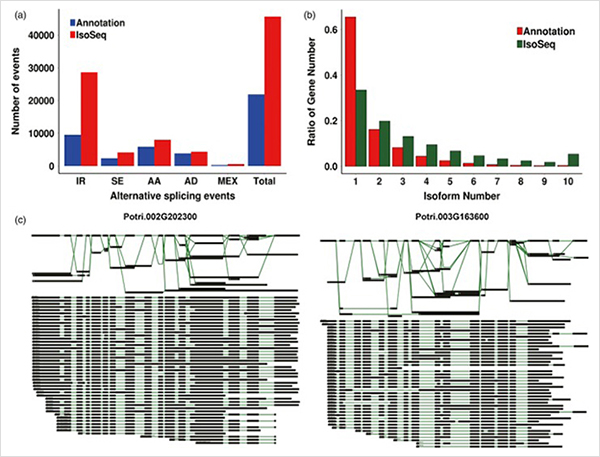
Analyse WGCNA sur les facteurs de transcription
Référence
Chao Q, Gao ZF, Zhang D et al.La dynamique de développement du transcriptome de la tige de Populus.Plant Biotechnol J.2019;17(1):206-219.est ce que je:10.1111/pbi.12958





