

Génomique comparée






Avantages des services
● Package d'analyses complet, contenant les huit analyses les plus couramment requises
● Haute fiabilité de l'analyse avec une interprétation détaillée et facilement compréhensible des résultats
● Chiffres bien conçus et prêts à publier
● Une équipe bioinformatique hautement qualifiée répond à diverses demandes d'analyse personnalisée
● Délai d'exécution plus court avec une plus grande précision d'analyse
● Une expérience abondante avec plus de 90 cas réussis avec un facteur d'impact cumulé publié de plus de 900
Spécifications des services
| Délai d'exécution estimé | Nombre d'espèces | Analyses |
| 30 jours ouvrables | 6 - 12 | Regroupement de familles de gènes Expansion et contraction de la famille de gènes Construction d'arbres phylogénétiques Estimation du temps de divergence (étalonnage fossile requis) Temps d'insertion LTR (Pour les plantes) Duplication du génome entier (pour les plantes) Pression sélective Analyse de synthèse |
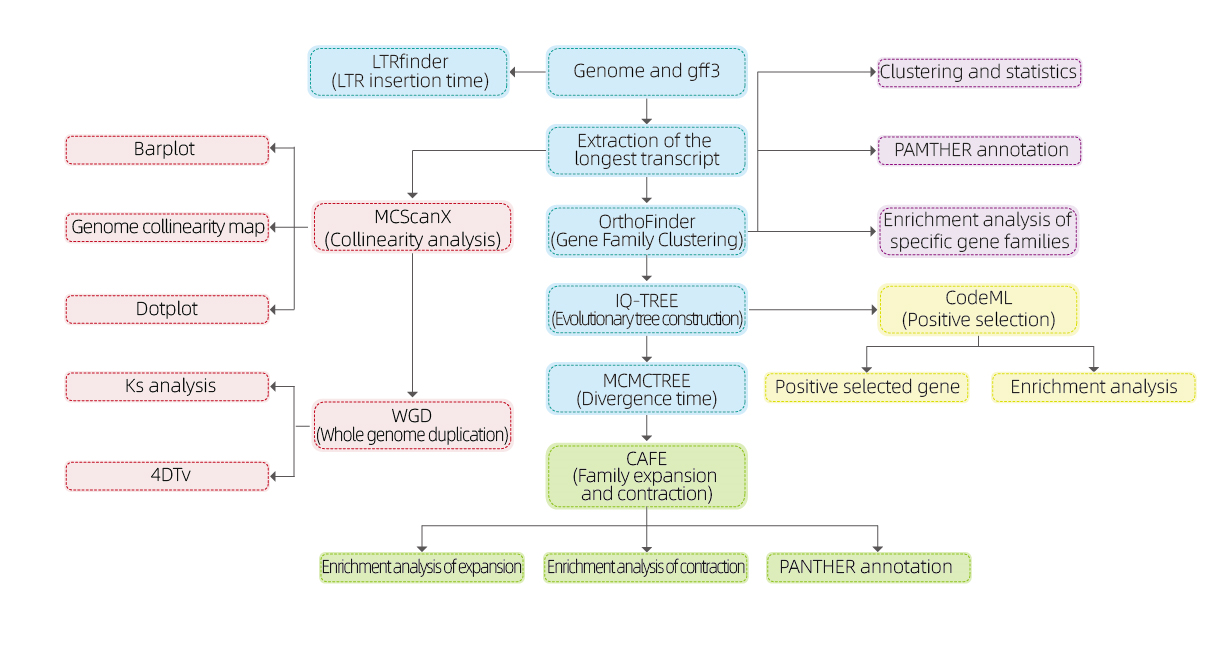
Analyses bioinformatiques
● Famille de gènes
● Phylogénétique
● Temps de divergence
● Pression sélective
● Analyse de synthèse
Exigences et livraison des échantillons
Exigences de l'échantillon :
Tissus ou ADN pour le séquençage et l'assemblage du génome
Pour les tissus
| Espèces | Tissu | Enquête | PacBio CCS |
| Animal | Tissu viscéral | 0,5 ~ 1g | ≥ 3,5g |
| Tissu musculaire | |||
| ≥ 5,0g | |||
| ≥ 5,0 ml | |||
| Sang de mammifère | |||
| ≥ 0,5 ml | |||
| Sang de volaille/poisson | |||
| Usine | Feuille fraîche | 1 ~ 2g | ≥ 5,0g |
| Pétale/Tige | 1 ~ 2g | ≥ 10,0g | |
| Racine/graine | 1 ~ 2g | ≥ 20,0g | |
| Cellules | Cellule cultivée | - | ≥ 1 x 108 |
Données
Fichiers de séquences génomiques (.fasta) et fichiers d'annotation (.gff3) d'espèces étroitement apparentées
Flux de travail des services

Conception d'expériences

Livraison d'échantillon

Construction d'une bibliothèque

Séquençage

L'analyse des données

Services après-vente
*Les résultats de démonstration présentés ici proviennent tous de génomes publiés avec Biomarker Technologies
1.Estimation du temps d'insertion des LTR : la figure montre une distribution bimodale unique des temps d'insertion des LTR-RT dans le génome du seigle Weining, par rapport à d'autres espèces.Le pic le plus récent est apparu il y a environ 0,5 million d’années.
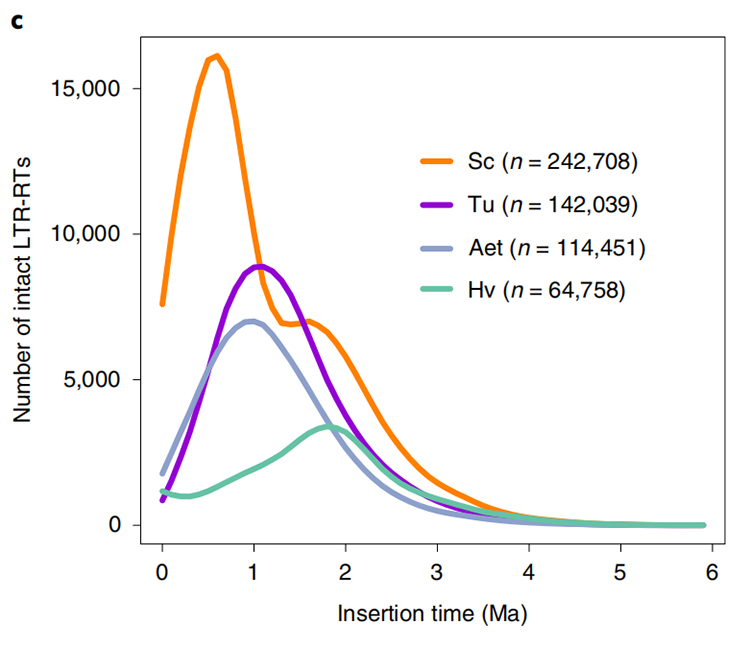
Li Guang et coll.,Génétique naturelle, 2021
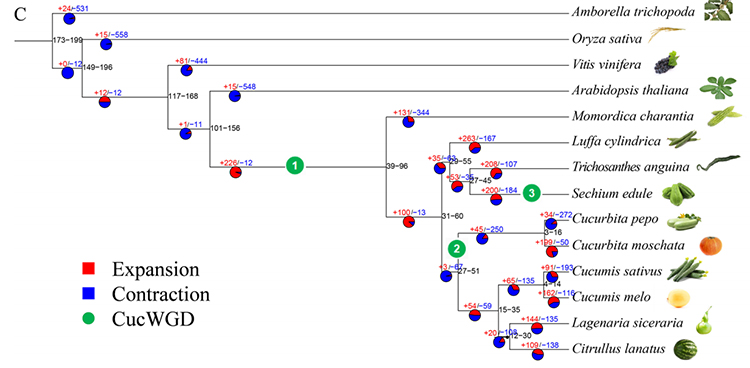
2.Phylogénie et analyse de la famille génétique de la chayote (Sechium edule) : En analysant la chayote et les 13 autres espèces apparentées de la famille des gènes, la chayote s'est révélée être la plus étroitement apparentée à la courge serpent (Trichosanthes anguina).La chayote dérivée de la courge serpent vers 27-45 Mya et la duplication du génome entier (WGD) ont été observées chez la chayotte dans 25 ± 4 Mya, ce qui est le troisième événement WGD chez les cucuibitaceae.
Fu A et coll.,Recherche horticole, 2021
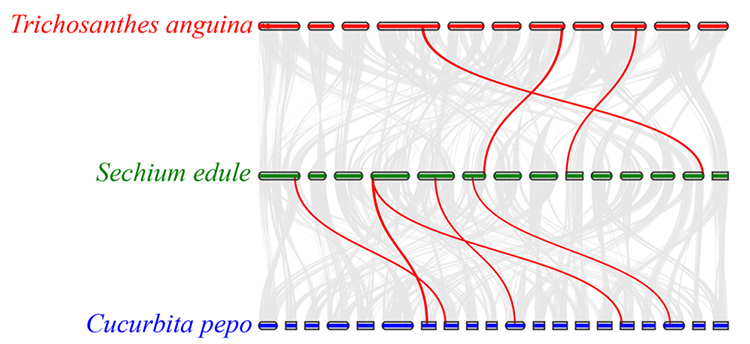
3. Analyse de synthèse : certains gènes liés aux phytohormones dans le développement des fruits ont été trouvés dans la chayote, la courge serpent et la courge.La corrélation entre la chayote et la courge est légèrement supérieure à celle entre la chayote et la courge serpent.
Fu A et coll.,Recherche horticole, 2021
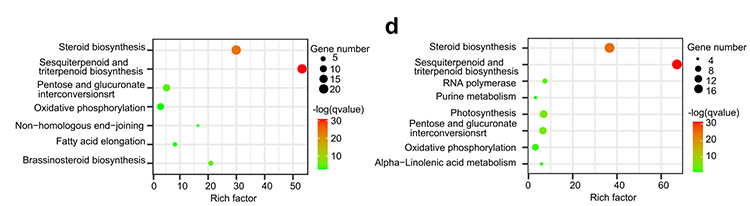
4. Analyse de la famille de gènes : l'enrichissement KEGG sur l'expansion et la contraction de la famille de gènes dans les génomes de G.thurberi et G.davidsonii a montré que les gènes liés à la biosynthèse des stéroïdes et des brassinostéroïdes étaient élargis.
Yang Z et coll.,Biologie BMC, 2021
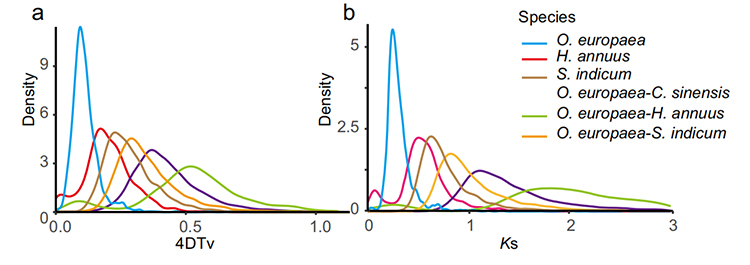
5. Analyse de duplication du génome entier : l'analyse de la distribution 4DTV et Ks a montré un événement de duplication du génome entier.Les pics intraspécifiques ont montré des événements de duplication.Les pics interspécifiques montrent des événements de spéciation.L'analyse a indiqué que, comparée aux trois autres espèces étroitement apparentées, O. europaea a récemment subi une duplication génétique à grande échelle.
Rao G et coll.,Recherche horticole, 2021
Affaire BMK
Rose sans piquant : connaissances génomiques liées à l'adaptation à l'humidité
Publié : Revue scientifique nationale, 2021
Stratégie de séquençage :
'Basye'sSans épines" (R.Wichurainan) génome :
Environ.93 X PacBio + env.90 X Nanopores + 267 X Illumina
Résultats clés
1. Le génome de R.wichuraiana de haute qualité a été construit à l'aide de techniques de séquençage à lecture longue, qui donnent un assemblage de 530,07 Mo (la taille estimée du génome était d'environ 525,9 Mo par cytométrie en flux et 525,5 par enquête génomique ; L'hétérozygotie était d'environ 1,03 %).Le score estimé par BUSCO était de 93,9 %.En comparaison avec « Old Blush » (haploOB), la qualité et l'exhaustivité de ce génome ont été confirmées par la précision de base unique et l'indice d'assemblage LTR (LAI = 20,03).Le génome de R.wichuraiana contient 32 674 gènes codant pour des protéines.
2. Une analyse conjointe multi-omique, comprenant la génomique comparative, la transcriptomique et l'analyse QTL de la population génétique, a révélé la spéciation cruciale entre R. wichuraiana et Rosa chinensis.En outre, la variation de l'expression de gènes apparentés dans QTL était probablement associée à la configuration des piquants de la tige.
L'analyse génomique comparative entre Basye;s Thornless et Rosa chinensis, y compris l'analyse de la synténie, les groupes de familles de gènes, l'analyse de l'expansion et de la contraction, a révélé un grand nombre de variations liées à des caractères cruciaux des roses.L’expansion unique de la famille des gènes NAC et FAR1/FRS était très probablement associée à la résistance aux taches noires.
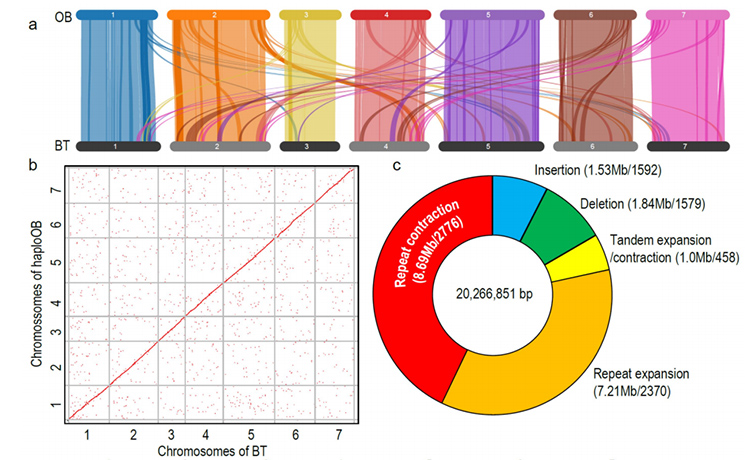

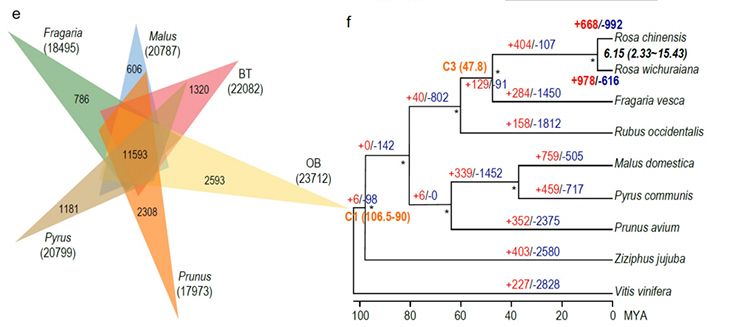
Analyse génomique comparative entre les génomes BT et haploOB.
Zhong, M. et coll.« Rose sans piquant : connaissances génomiques liées à l’adaptation à l’humidité »Revue scientifique nationale, 2021 ;, nwab092.





