

IHMISGENOMIIKKA
luonnon genetiikka
Pitkään luettu sekvensointi tunnistaa GGC-toistolaajennukset NOTCH2NLC:ssä, jotka liittyvät hermosolujen intranukleaariseen inkluusiotautiin
ONT uudelleensekvensointi |Illumina |Koko eksomin sekvensointi |CRISPR-Cas9 ONT kohdistettu sekvensointi |RNA-seq |ONT 5mC metylaatiokutsu
Kohokohdat
1. Suuren NIID-perheen linkitysanalyysillä tunnistettiin kaksi yhdistettyä aluetta.
2.ONT-pohjainen pitkälukuinen sekvensointi ja Cas-9-välitteinen rikastus ONT-sekvensointi havaitsi mahdollisen geneettisen syyn NIID-, GGC-toistolaajennuksiin NOTCH2NLC:n 5′ UTR:ssä.Tämä tutkimus raportoi toistuvista laajennuksista ihmisspesifisissä geeneissä ensimmäistä kertaa, jotka kehittyivät segmentaalisten päällekkäisyyksien kautta.
3. RNA-sekvensointi paljasti epänormaaleja antisense-transkriptejä NOTCH2NLC:n GGC-toiston laajennusalueiden alussa tai sisällä.
Tausta
Neuronal intranukleaarinen inkluusiotauti (NIID) on etenevä ja kuolemaan johtava neurodegeneratiivinen sairaus, jolle on tunnusomaista eosinofiilisten hyaliinisten intranukleaaristen inkluusioiden esiintyminen keskus- ja ääreishermostossa.Sen erittäin vaihtelevat kliiniset ilmenemismuodot aiheuttavat suuria vaikeuksia diagnosoinnissa ihobiopsian käyttöönottoon asti.Histopatologiaan perustuvat menetelmät kärsivät kuitenkin edelleen virhediagnoosista, mikä vaatii NIID:n geneettistä ymmärtämistä.
Saavutukset
Kytkentäanalyysi
Short-read-sekvensointiin perustuva koko genomin sekvensointi (WGS) ja koko eksomin sekvensointi (WES) suoritettiin suurelle NIID-perheelle (13 sairasta ja 7 ei-sairaista jäsentä).Näistä tiedoista poimittujen SNP:iden kytkentäanalyysi paljasti vain kaksi linkitettyä aluetta: 3,5 Mb:n alueen kohdissa 1p36,31-p36,22 (maksimi LOD = 2,32) ja 58,1 Mb:n alueen kohdissa 1p22,1-q21,3 (maksimi LOD: 4,21). ).Näillä linkitetyillä alueilla ei kuitenkaan tunnistettu patogeenisiä SNP:itä tai CNV:itä.
GGC toistaa laajennuksia NOTCH2NLC:ssä
Nanopore-pohjainen sekvensointi käsiteltiin 13:lla sairastuneella ja 4:llä vahingoittumattomalla jäsenellä 8 perheestä (toinen sairastunut jäsen sekvensoitiin Pacbio long read -sekvensointialustalla.).Pitkään luetut tiedot paljastivat sairauteen liittyviä GGC-toistolaajennuksia NOTCH2NLC-geenin 5' UTR:ssä, joka kartoitettiin 58,1 Mb:n linkitetylle alueelle (kuva 1).Nämä toistuvat laajennukset tunnistettiin myös kaikissa 40 satunnaisessa NIID-tapauksessa, jotka testattiin RP-PCR:llä.
Cas-9-välitteistä kohdesekvensointia nanopore-alustalla käytettiin korkeamman lukupeiton saavuttamiseksi NOTCH2NLC-toistolla (100 X-1 795 X).Nämä konsensussekvenssit sopivat hyvin aikaisempien GGC-toistolaajennusten havaintojen kanssa.Lisäksi {(GGA)n(GGC)n}n toistot tunnistettiin potentiaaliseksi geneettiseksi markkeriksi heikkousdominoivalle fenotyypille (kuvio 2).
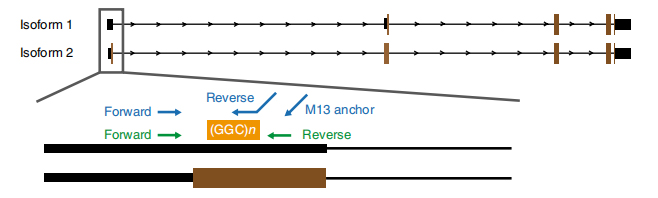
Kuva 1. Tautiin liittyvä toistuva laajeneminen, joka tunnistettiin NOTCH2NLC-isoformien eksonissa 1.
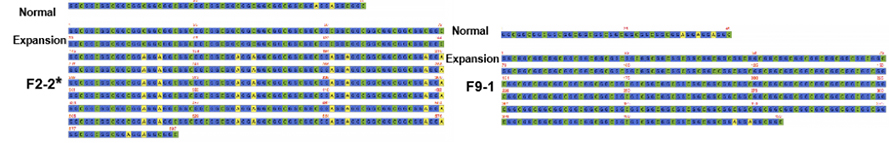
Kuva 2. NPTCH2NLC-toiston konsensussekvenssit NIID-potilailla, joilla on (*) tai ei ole heikkousdominoiva fenotyyppi
NOTCH2NL-geenit ovat ihmisspesifisiä geenejä, joilla uskotaan olevan tärkeä rooli ihmisen aivojen evoluutiossa ja neurologisissa sairauksissa.Kolme NOTCH2:een liittyvää geeniä (NOTCH2NLA, NOTCH2NLB ja NOTCH2NLC), joiden sekvenssi-identtisyys on >99,1 %, ei kuitenkaan ratkaistu ennen viimeisintä ihmisen genomikokoonpanoa.Synteesivapaa ja pitkälukuinen sekvensointi nanohuokosalustalla on osoittanut merkittäviä etuja erittäin samankaltaisten alueiden ja (GGC)n toistojen ratkaisemisessa 100 % GC-rikkailla alueilla.
GGC toistaa laajennuksia NOTCH2NLC:ssä
Transcriptome-sekvensointi käsiteltiin kahdella sairastuneella ja kahdella vahingoittumattomalla jäsenellä.Normalisoitu lukusyvyys laskettiin sense- ja antisense-säikeistä ylävirtaan NOTCH2NL-paralogien ensimmäisistä eksoneista.Epänormaalit antisense-transkriptit löydettiin vain sairastuneissa tapauksissa, jotka sijaitsevat toistuvan laajennusalueen alussa tai sisällä (Purple piikit F1-14:ssä ja F1-16:ssa kuvassa 3.).Lisäksi tunnistettiin 54 DEG:tä ja kaikki olivat rikastettuja hermosolujen toimintoihin liittyvillä GO- ja MPO-termeillä.
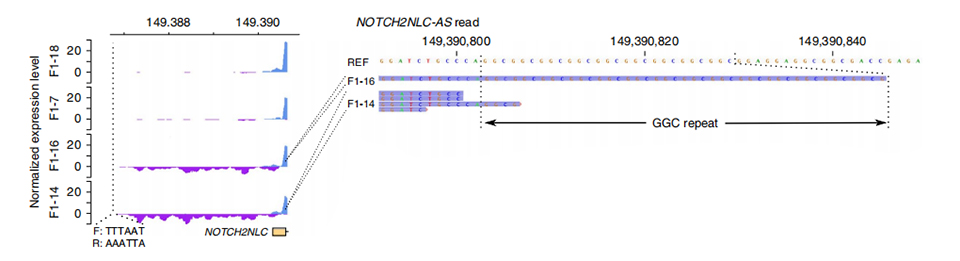
Kuva 3. Normalisoitu lukusyvyys NOTCH2NLC:n ensimmäisestä eksonista vastavirtaan vaikuttamattomissa (yllä) ja vaikutuksen alaisina (alla) tapauksissa.
Tekniikka
Oxford Nanopore Teghnologies (ONT)
Nanopore-sekvensointi erottuu muista sekvensointialustoista siinä, että nukleotidit luetaan suoraan ilman DNA-synteesiprosessia.Kun yksijuosteinen DNA kulkee nanokokoisen proteiinihuokosen (nanohuokosen) läpi, eri nukleotidit tuottavat erilaisia ionivirtoja, jotka voidaan siepata ja siirtää emässekvenssiin.ONT-sekvensointialusta itsessään ei osoita ilmeistä teknistä rajaa DNA-lukemisen pituudelle.Siksi ultra-long reads (ULR) on saatavilla korkealaatuiseen genomin kokoamiseen.Lisäksi nämä erittäin pitkät lukemat, jotka ovat riittävän pitkiä ylittämään monimutkaisia sekvenssiominaisuuksia tai rakenteellisia vaihteluita, auttavat voittamaan lyhyen lukujakson rajoitukset tässä.

Nanopore-sekvensointi
Rakennevaihteluiden (SV) tunnistaminen
Ssynteesivapaa sekvensointi säilytti suurelta osin DNA-metylaatiotiedot templaatissa.Metyloitu A, T, C ja G tuottavat erillisiä ionivirtoja metyloimattomista, jotka alusta voi lukea suoraan.Nanohuokosekvensointi mahdollistaa sekä 5 mC:n että 6 mA:n koko genomin profiloinnin yhden nukleotidin resoluutiolla.
Viite
Jun Sone, et.al.Pitkään luettu sekvensointi tunnistaa GGC-toistolaajennukset NOTCH2NLC:ssä, jotka liittyvät hermosolujen intranukleaariseen inkluusiotautiin.Nature Genetics (2019)
Tekniikka ja kohokohdat Tavoitteena on jakaa erilaisten korkean suorituskyvyn sekvensointiteknologioiden viimeisimpiä onnistuneita sovelluksia eri tutkimusareenoilla sekä loistavia ideoita kokeelliseen suunnitteluun ja tiedon louhintaan.
Postitusaika: 06.01.2022