

توالی mRNA مبتنی بر غیر مرجع-Illumina








امکانات
● مستقل از هر ژنوم مرجع،
● داده ها می توانند برای تجزیه و تحلیل ساختار و بیان رونوشت ها استفاده شوند
● سایت های برش متغیر را شناسایی کنید
مزایای خدمات
● تحویل نتایج مبتنی بر BMKCloud: نتایج بهعنوان فایل داده و گزارش تعاملی از طریق پلتفرم BMKCloud ارائه میشوند، که امکان خواندن کاربرپسند خروجیهای تحلیل پیچیده و دادهکاوی سفارشیشده بر اساس تجزیه و تحلیل بیوانفورماتیک استاندارد را فراهم میکند.
● خدمات پس از فروش: خدمات پس از فروش با اعتبار 3 ماه پس از اتمام پروژه شامل پیگیری پروژه ها، عیب یابی، پرسش و پاسخ نتایج و غیره.
نمونه مورد نیاز و تحویل
نمونه مورد نیاز:
نوکلئوتیدها:
| Conc.(ng/μl) | مقدار (μg) | خلوص | تمامیت |
| ≥ 20 | ≥ 0.5 | OD260/280=1.7-2.5 OD260/230=0.5-2.5 آلودگی پروتئین یا DNA محدود یا بدون آلودگی روی ژل نشان داده شده است. | برای گیاهان: RIN≥6.5; برای حیوانات: RIN≥7.0; 5.0≥28S/18S≥1.0; ارتفاع پایه محدود یا بدون ارتفاع |
بافت: وزن (خشک): ≥1 گرم
*برای بافت کوچکتر از 5 میلی گرم، توصیه می کنیم نمونه بافت منجمد شده (در نیتروژن مایع) را ارسال کنید.
سوسپانسیون سلولی: تعداد سلول = 3×107
*ما توصیه می کنیم لیز سلولی منجمد را ارسال کنید.در صورتی که تعداد آن سلول کوچکتر از 10×5 باشد5، منجمد فلاش در نیتروژن مایع توصیه می شود.
نمونه های خون:
PA×geneBloodRNATube;
6mLTRIZol و 2mL خون (TRIzol:Blood=3:1)
تحویل نمونه توصیه شده
ظرف:
لوله سانتریفیوژ 2 میلی لیتری (فیل قلع توصیه نمی شود)
برچسب زدن نمونه: گروه + تکرار به عنوان مثال A1، A2، A3.B1، B2، B3...
حمل و نقل:
1. یخ خشک: نمونه ها باید در کیسه های بسته بندی شده و در یخ خشک دفن شوند.
لوله های پایدار RNA: نمونه های RNA را می توان در لوله تثبیت کننده RNA (به عنوان مثال RNAstable®) خشک کرد و در دمای اتاق حمل کرد.
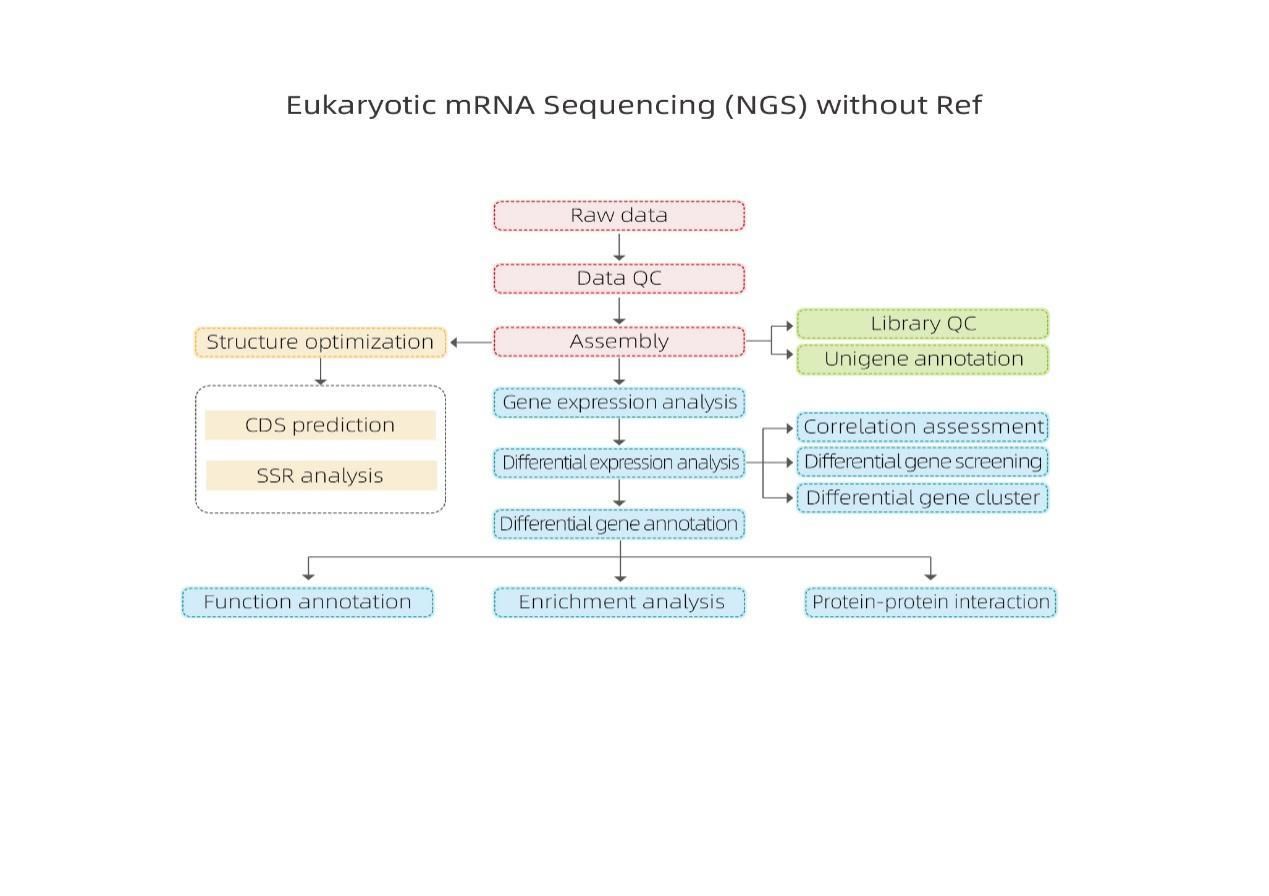
جریان کار خدمات

طراحی آزمایش

تحویل نمونه

استخراج RNA

ساخت کتابخانه

ترتیب دهی

تحلیل داده ها

خدمات پس از فروش
بیوانفورماتیک
1.mRNA(denovo) اصل مونتاژ
توسط Trinity، خوانده ها به قطعات کوچکتر، معروف به K-mer تقسیم می شوند.این K-merها سپس به عنوان دانه برای گسترش به contigs و سپس جزء بر اساس همپوشانی contig استفاده می شود.در نهایت، De Bruijn در اینجا برای شناسایی رونوشت ها در مؤلفه ها اعمال شد.
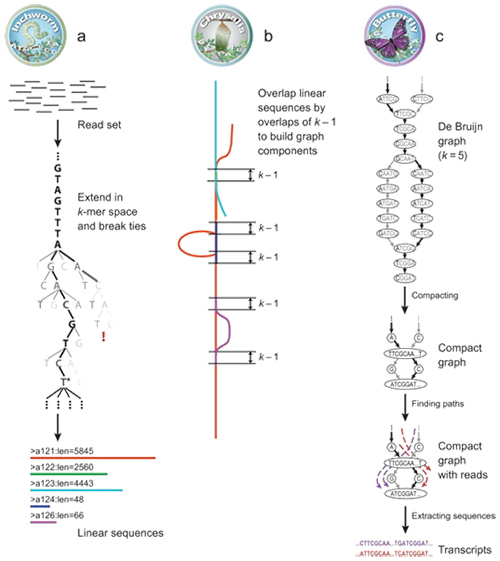
mRNA (De novo) بررسی اجمالی ترینیتی
2.mRNA (De novo) توزیع سطح بیان ژن
RNA-Seq قادر به دستیابی به یک تخمین بسیار حساس از بیان ژن است.به طور معمول، محدوده قابل تشخیص رونوشت عبارت FPKM از 10^-2 تا 10^6 متغیر است.
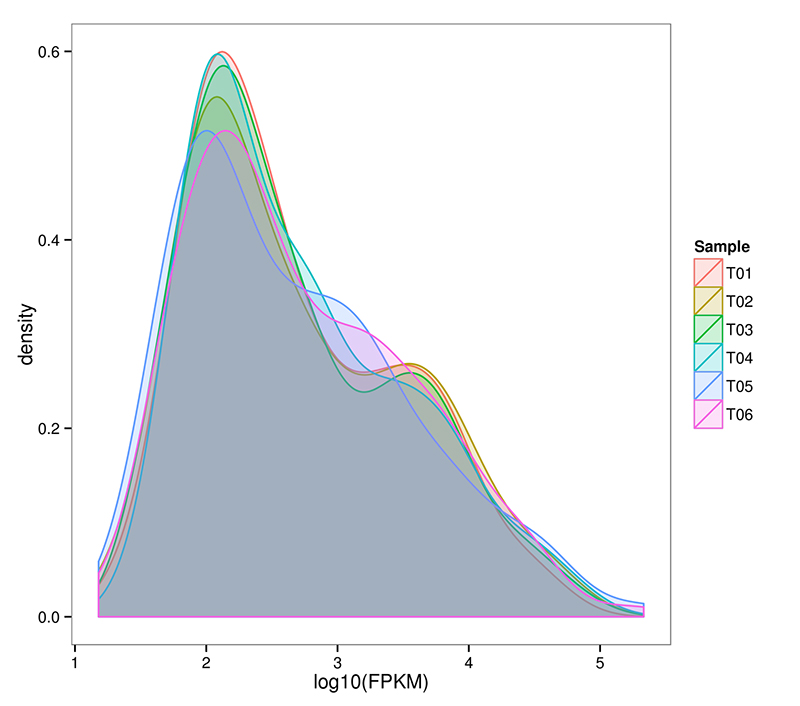
mRNA (De novo) توزیع چگالی FPKM در هر نمونه
3.mRNA (De novo) تجزیه و تحلیل GO غنی سازی DEGs
پایگاه داده GO (Gene Ontology) یک سیستم حاشیه نویسی بیولوژیکی ساختار یافته است که شامل واژگان استانداردی از عملکردهای ژن و محصولات ژنی است.این شامل چندین سطح است، که در آن هر چه سطح پایین تر باشد، عملکردها خاص تر هستند.
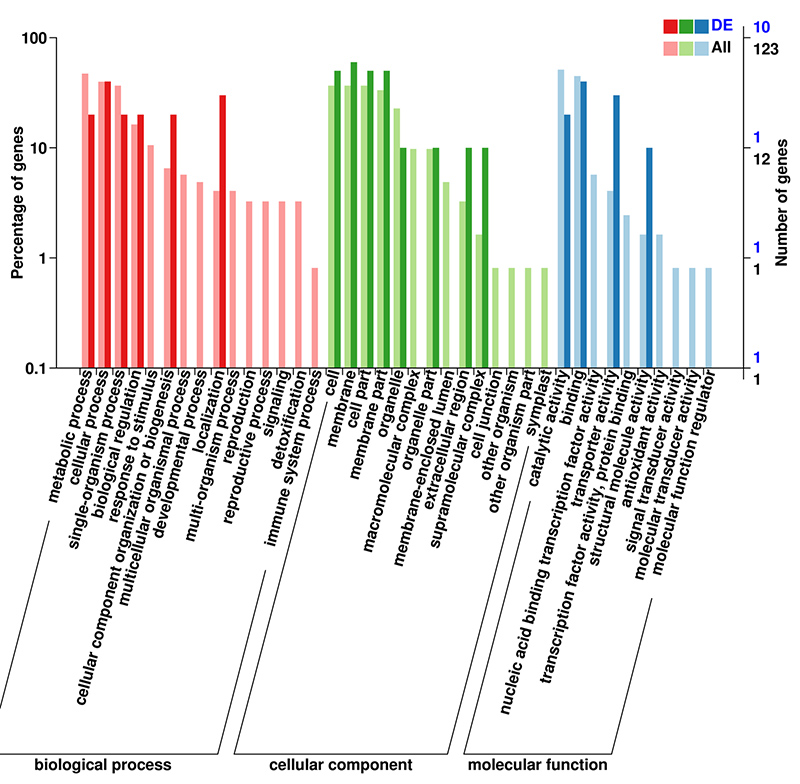
mRNA (De novo) GO طبقه بندی DEG ها در سطح دوم
کیس BMK
تجزیه و تحلیل رونوشت متابولیسم ساکارز در طول تورم و توسعه پیاز در پیاز (Allium cepa L.)
منتشر شده: مرزها در علوم گیاهی2016
استراتژی توالی
ایلومینا HiSeq2500
مجموعه نمونه
در این مطالعه از رقم یوتا یلو سوئیت اسپانیا "Y1351" استفاده شد.تعداد نمونه های جمع آوری شده بود
پانزدهمین روز پس از تورم (DAS) لامپ (قطر 2 سانتی متر و وزن 3-4 گرم)، 30مین DAS (قطر 5 سانتی متر و وزن 100-110 گرم) و حدود 3 در DAS 40 (قطر 7 سانتی متر و 260-300 گرم).
نتایج کلیدی
1. در نمودار ون، در مجموع 146 DEG در هر سه جفت مرحله رشد شناسایی شد.
2. "انتقال و متابولیسم کربوهیدرات" تنها با 585 یونیژن (یعنی 7٪ از COG مشروح شده) نشان داده شد.
3. Unigenes با موفقیت در پایگاه داده GO به سه دسته اصلی برای سه مرحله مختلف توسعه لامپ طبقه بندی شدند.بیشتر در دسته اصلی "فرایند بیولوژیکی" "فرایند متابولیک" و پس از آن "فرایند سلولی" بودند.در مقوله اصلی "عملکرد مولکولی" دو مقوله که بیشترین نمایش را دارند عبارتند از "پیوند" و "فعالیت کاتالیزوری".
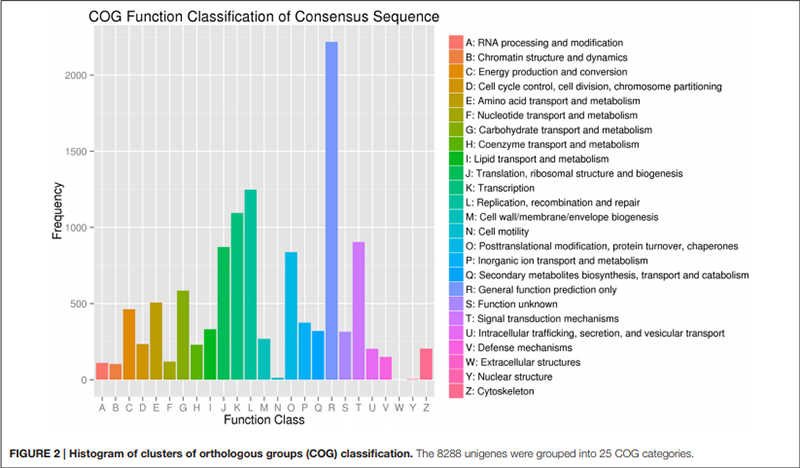 طبقه بندی هیستوگرام خوشه های گروه های ارتولوگ (COG). | 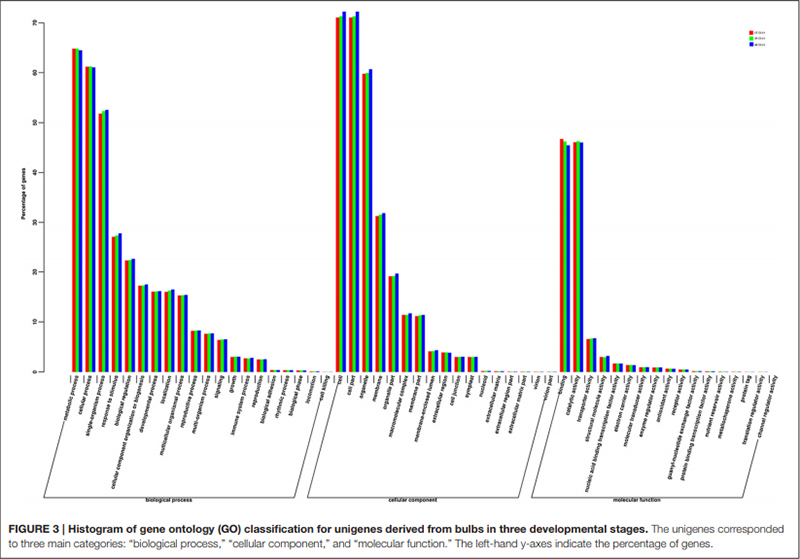 هیستوگرام طبقه بندی هستی شناسی ژن (GO) برای یونیژن های مشتق شده از لامپ در سه مرحله رشد |
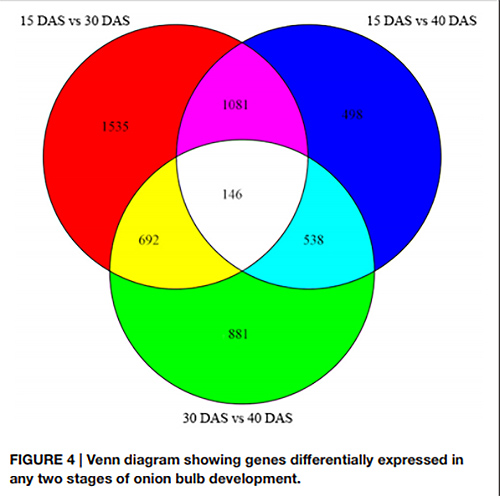 نمودار ون ژنهایی را نشان میدهد که در هر دو مرحله از رشد پیاز به طور متفاوت بیان میشوند |
ارجاع
Zhang C، Zhang H، Zhan Z، و همکاران.تجزیه و تحلیل رونوشت متابولیسم ساکارز در طول تورم و توسعه پیاز در پیاز (Allium cepa L.) [J].مرزها در علوم گیاهی، 2016، 7:1425-.DOI: 10.3389/fpls.2016.01425





