

Genética evolutiva








Ventajas del servicio
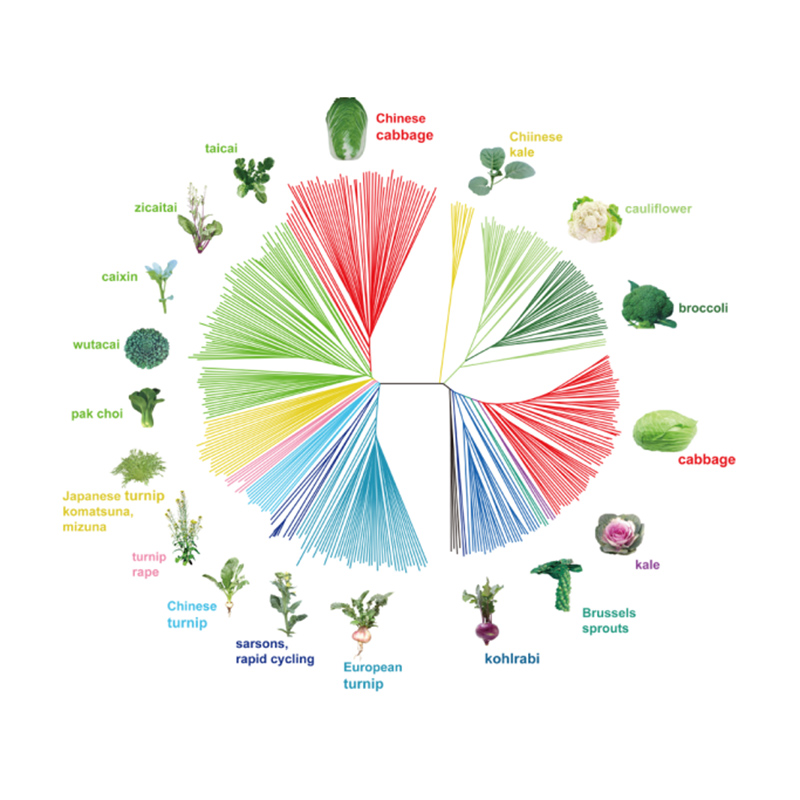
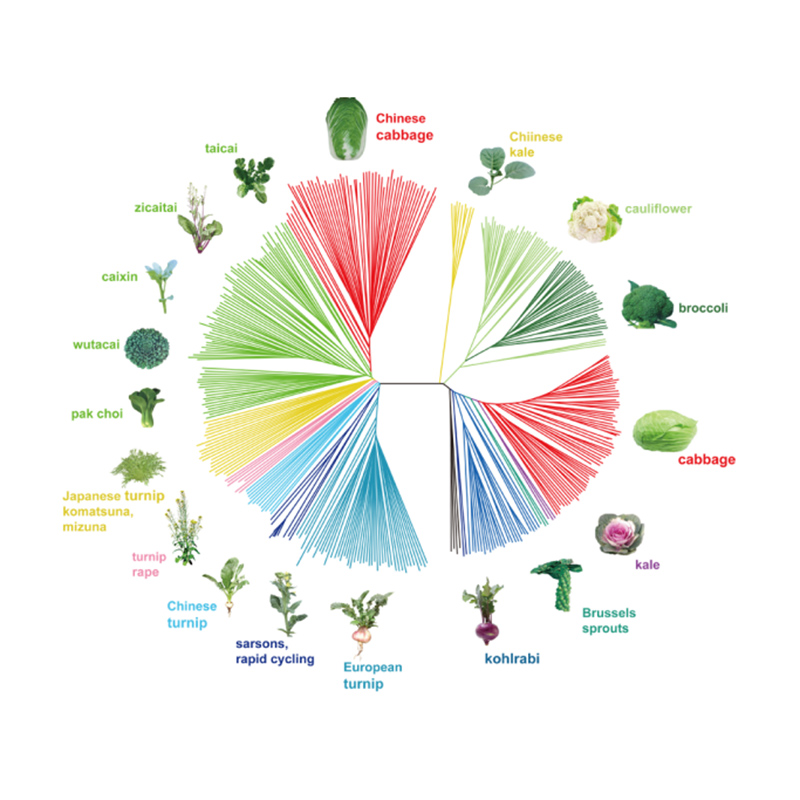
Takagi et al.,El diario de las plantas, 2013
● Estimar el tiempo y la velocidad de divergencia de especies en función de variaciones a nivel de nucleótidos y aminoácidos
● Revelación de una relación filogenética más confiable entre especies con una influencia minimizada de la evolución convergente y la evolución paralela
● Construir vínculos entre cambios genéticos y fenotipos para descubrir genes relacionados con rasgos
● Estimar la diversidad genética, que refleja el potencial evolutivo de las especies.
● Tiempo de respuesta más rápido
● Amplia experiencia: BMK ha acumulado una enorme experiencia en proyectos relacionados con la población y la evolución durante más de 12 años, cubriendo cientos de especies, etc. y ha contribuido en más de 80 proyectos de alto nivel publicados en Nature Communications, Molecular Plants, Plant Biotechnology Journal, etc.
Especificaciones de servicio
Materiales:
Normalmente, se recomiendan al menos tres subpoblaciones (por ejemplo, subespecies o cepas).Cada subpoblación debe contener no menos de 10 individuos (plantas >15, puede reducirse para especies raras).
Estrategia de secuenciación:
* WGS se puede emplear para especies con genoma de referencia de alta calidad, mientras que SLAF-Seq es aplicable a especies con o sin genoma de referencia, o genoma de referencia de mala calidad.
| Aplicable al tamaño del genoma. | WGS | Etiquetas SLAF (×10.000) |
| ≤ 500 MB | 10×/individual | WGS es más recomendable |
| 500 MB - 1 GB | 10 | |
| 1GB - 2GB | 20 | |
| ≥2 GB | 30 |
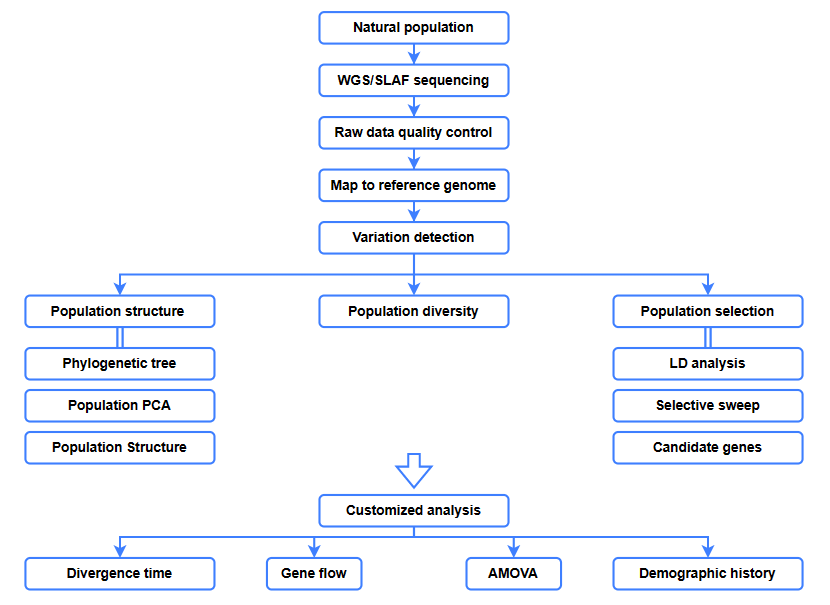
Análisis bioinformáticos.
● Análisis evolutivo
● Barrido selectivo
● Flujo genético
● Historia demográfica
● Tiempo de divergencia
Requisitos de muestra y entrega
Requisitos de muestra:
| Especies | Tejido | WGS-NGS | SLAF |
| Animal
| tejido visceral |
0,5~1g
|
0,5g
|
| Tejido muscular | |||
| sangre de mamífero | 1,5 ml
| 1,5 ml
| |
| Sangre de aves/pescado | |||
| Planta
| hoja fresca | 1~2g | 0,5~1g |
| Pétalo/tallo | |||
| Raíz/Semilla | |||
| Células | Célula cultivada |
| ADNg | Concentración | Cantidad (ug) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1,6-2,5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Flujo de trabajo del servicio

Diseño de experimentos

Entrega de muestra

construcción de biblioteca

Secuenciación

Análisis de los datos

Servicios postventa
*Los resultados de demostración que se muestran aquí provienen todos de genomas publicados con BMKGENE
1.El análisis de la evolución contiene la construcción de un árbol filogenético, una estructura poblacional y un PCA basados en variaciones genéticas.
El árbol filogenético representa las relaciones taxonómicas y evolutivas entre especies con un ancestro común.
PCA tiene como objetivo visualizar la cercanía entre subpoblaciones.
La estructura de la población muestra la presencia de subpoblaciones genéticamente distintas en términos de frecuencias alélicas.
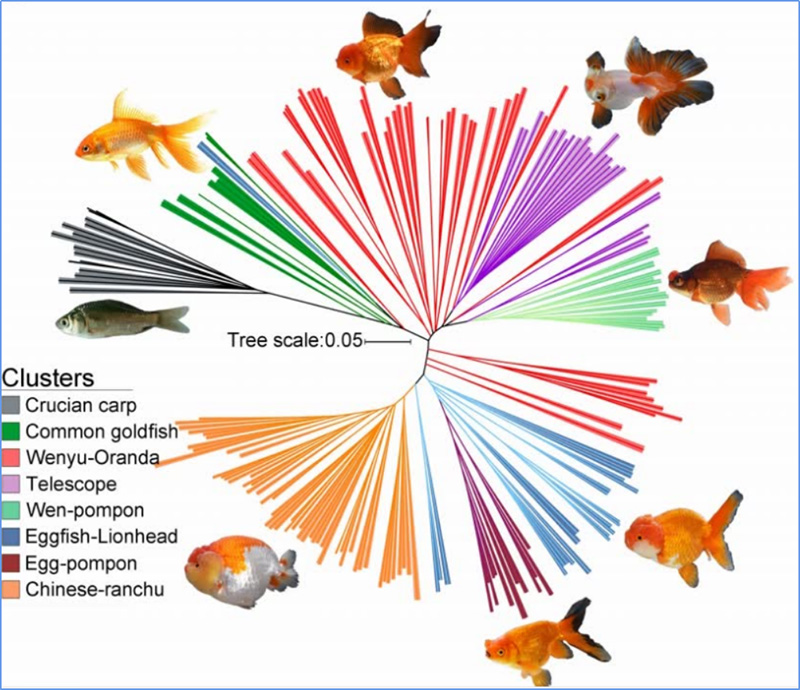
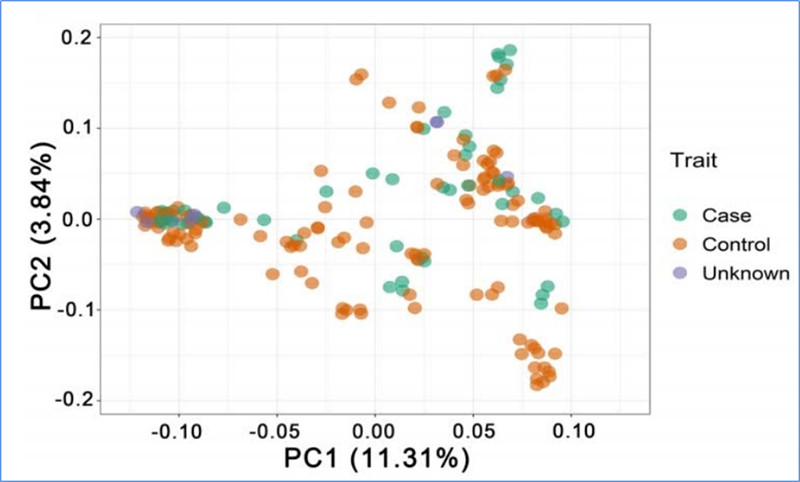
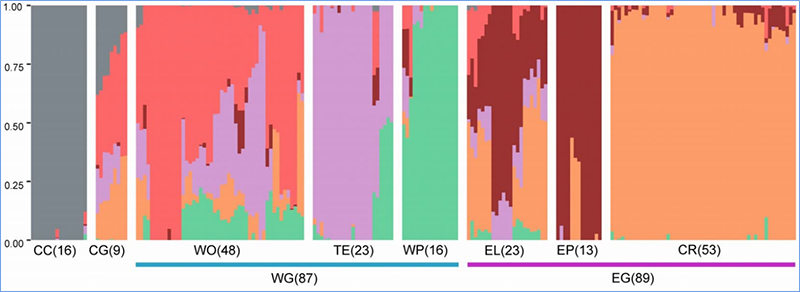
Chen, et.Alabama.,PNAS, 2020
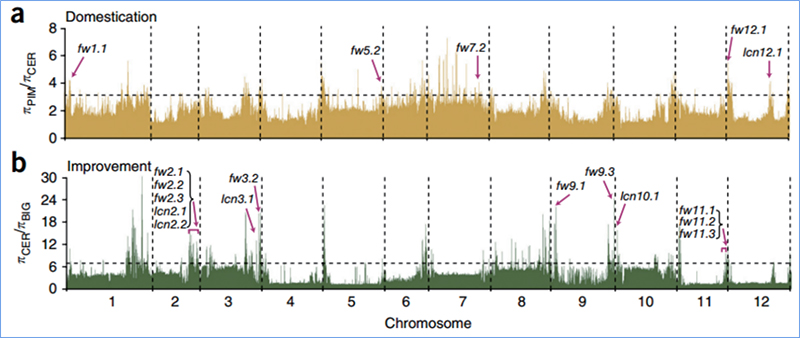
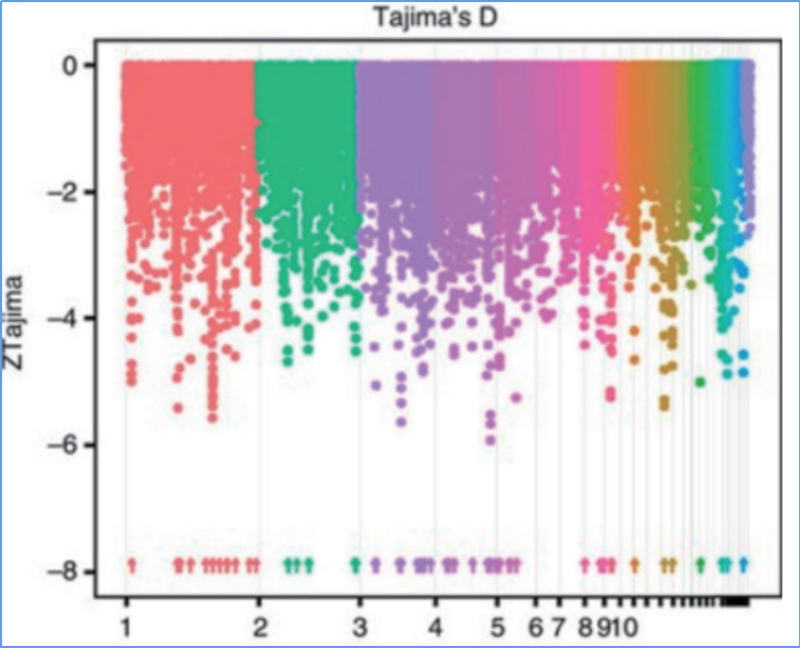
2.Barrido selectivo
El barrido selectivo se refiere a un proceso mediante el cual se selecciona un sitio ventajoso y las frecuencias de los sitios neutrales vinculados aumentan y las de los sitios no vinculados disminuyen, lo que resulta en una reducción de la regional.
La detección de todo el genoma en regiones de barrido selectivo se procesa calculando el índice genético de la población (π, Fst, D de Tajima) de todos los SNP dentro de una ventana deslizante (100 Kb) en un paso determinado (10 Kb).
Diversidad de nucleótidos (π)
D de Tajima
Índice de fijación (Fst)
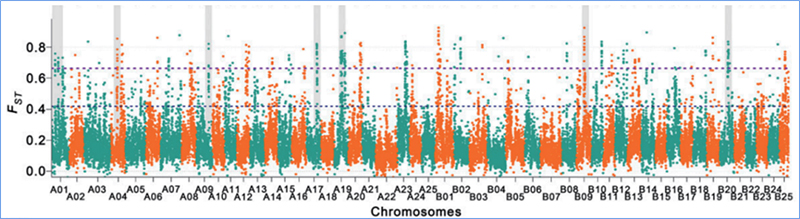
Wu, et.Alabama.,Planta molecular, 2018
3.Flujo de genes
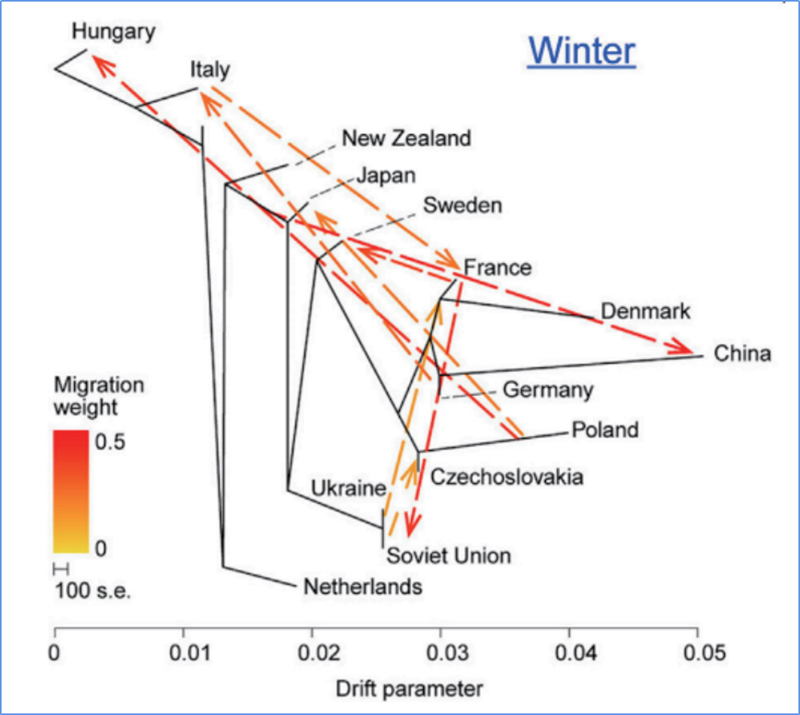
Wu, et.Alabama.,Planta molecular, 2018
4.Historia demográfica
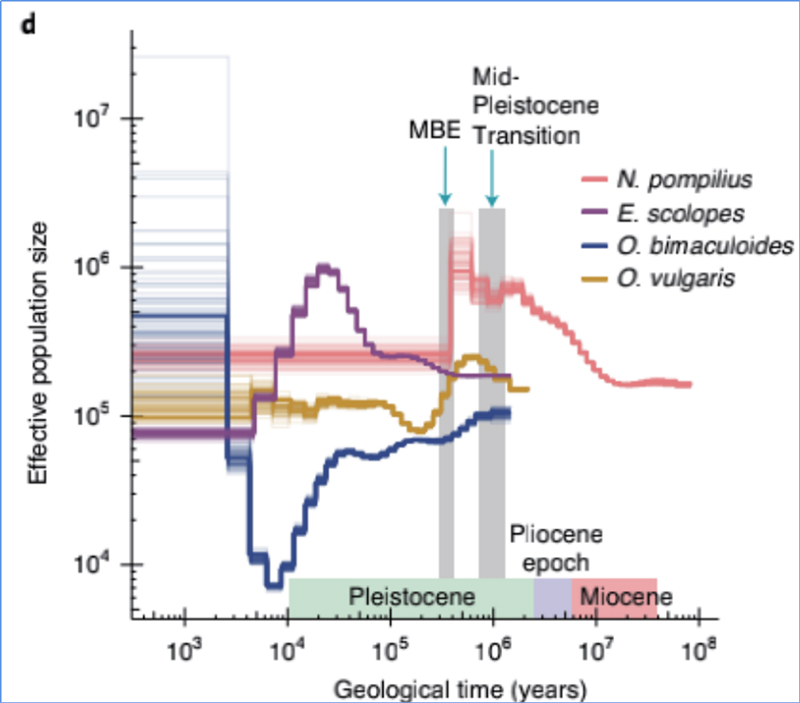
Zhang, et.Alabama.,Naturaleza Ecología y Evolución, 2021
5.Tiempo de divergencia
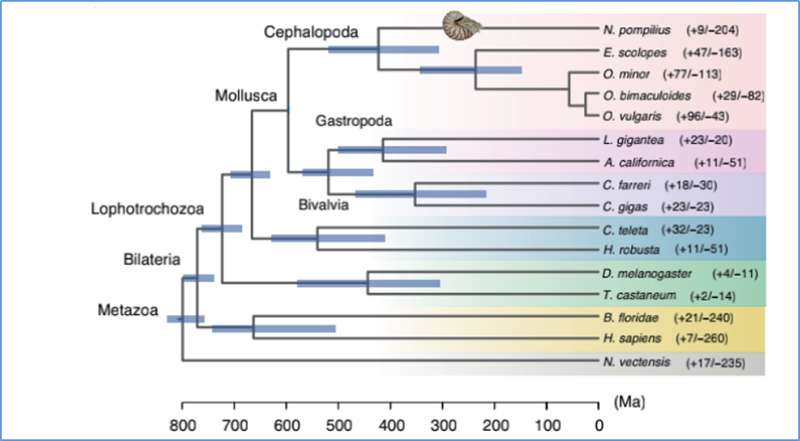
Zhang, et.Alabama.,Naturaleza Ecología y Evolución, 2021
Caso BMK
Un mapa de variación genómica proporciona información sobre la base genética de la selección de la col china de primavera (Brassica rapa ssp. Pekinensis)
Publicado: Planta molecular, 2018
Estrategia de secuenciación:
Resecuenciación: profundidad de secuenciación: 10×
Resultados clave
En este estudio, se procesaron 194 coles chinas para resecuenciarlas con una profundidad promedio de 10×, lo que arrojó 1.208.499 SNP y 416.070 InDels.El análisis filogenético de estas 194 líneas mostró que estas líneas se pueden dividir en tres ecotipos: primavera, verano y otoño.Además, la estructura poblacional y el análisis PCA indicaron que el repollo chino de primavera se originó a partir de un repollo de otoño en Shandong, China.Posteriormente se introdujeron en Corea y Japón, se cruzaron con líneas locales y algunas variedades de floración tardía se introdujeron en China y finalmente se convirtieron en repollo chino de primavera.
El escaneo de todo el genoma en coles chinas de primavera y coles de otoño en la selección reveló 23 loci genómicos que han pasado por una fuerte selección, dos de los cuales se superpusieron con la región de control del tiempo de floración basada en el mapeo de QTL.Se descubrió que estas dos regiones contienen genes clave que regulan la floración, BrVIN3.1 y BrFLC1.Se confirmó además que estos dos genes estaban involucrados en el tiempo de unión mediante estudios del transcriptoma y experimentos transgénicos.
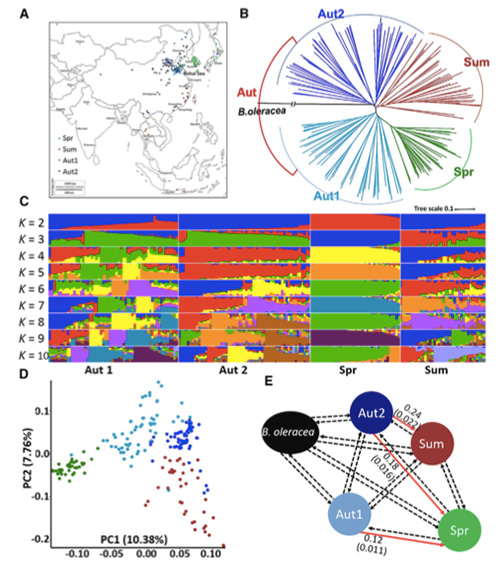 Análisis de la estructura poblacional de las coles chinas. | 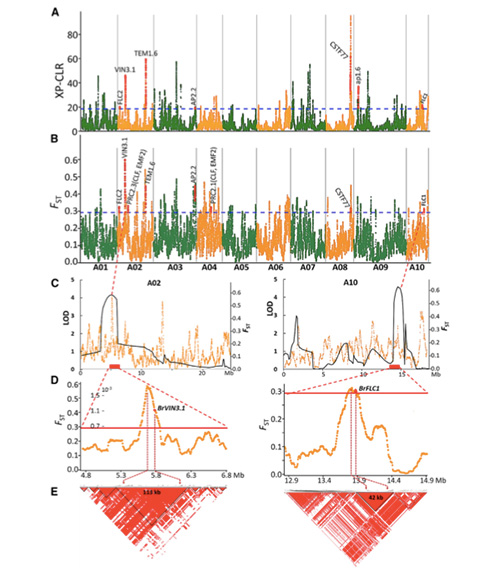 Información genética sobre la selección de col china. |
Tongbing, et al."Un mapa de variación genómica proporciona información sobre la base genética de la selección de repollo chino de primavera (Brassica rapa ssp.pekinensis)".plantas moleculares,11(2018):1360-1376.





