

Secuenciación de ARNm de longitud completa -PacBio





Ventajas del servicio
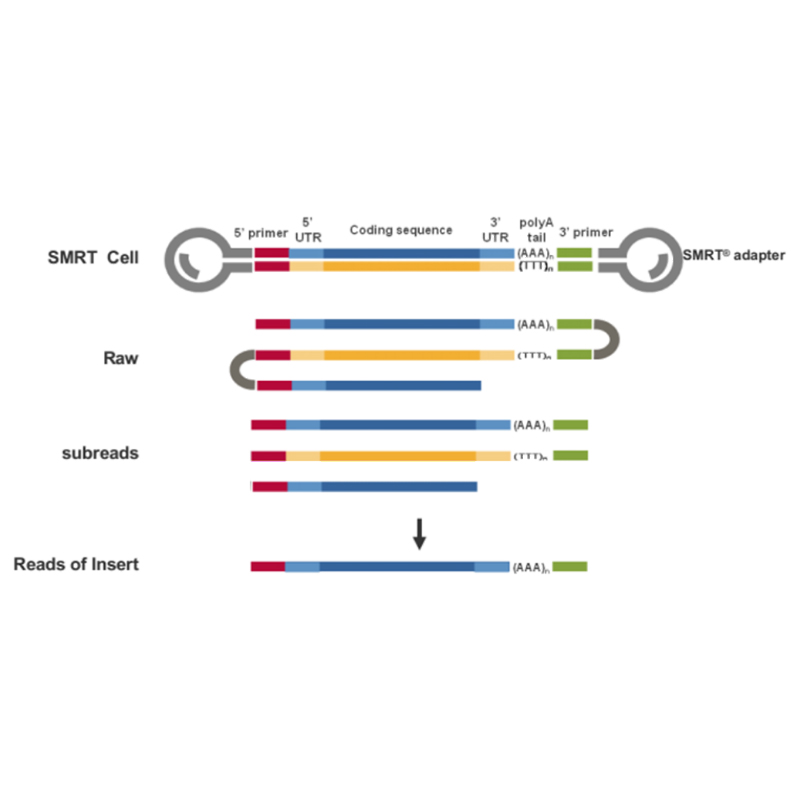
● Lectura directa de la molécula de ADNc de longitud completa desde el extremo 3' al extremo 5'
● Resolución de nivel de isoforma en estructura de secuencia
● Transcripciones con alta precisión e integridad.
● Altamente compatible con diversas especies.
● Gran capacidad de secuenciación con 4 plataformas de secuenciación PacBio Sequel II equipadas
● Alta experiencia con más de 700 proyectos de secuenciación de ARN basados en Pacbio
● Entrega de resultados basada en BMKCloud: minería de datos personalizada disponible en la plataforma.
● Servicios posventa válidos por 3 meses una vez finalizado el proyecto.
Especificaciones de servicio
Plataforma: PacBio Secuela II
Biblioteca de secuenciación: biblioteca de ARNm enriquecida con poli A
Rendimiento de datos recomendado: 20 Gb/muestra (Dependiendo de la especie)
FLNC(%): ≥75%
*FLNC: transcripciones completas no quiméricas
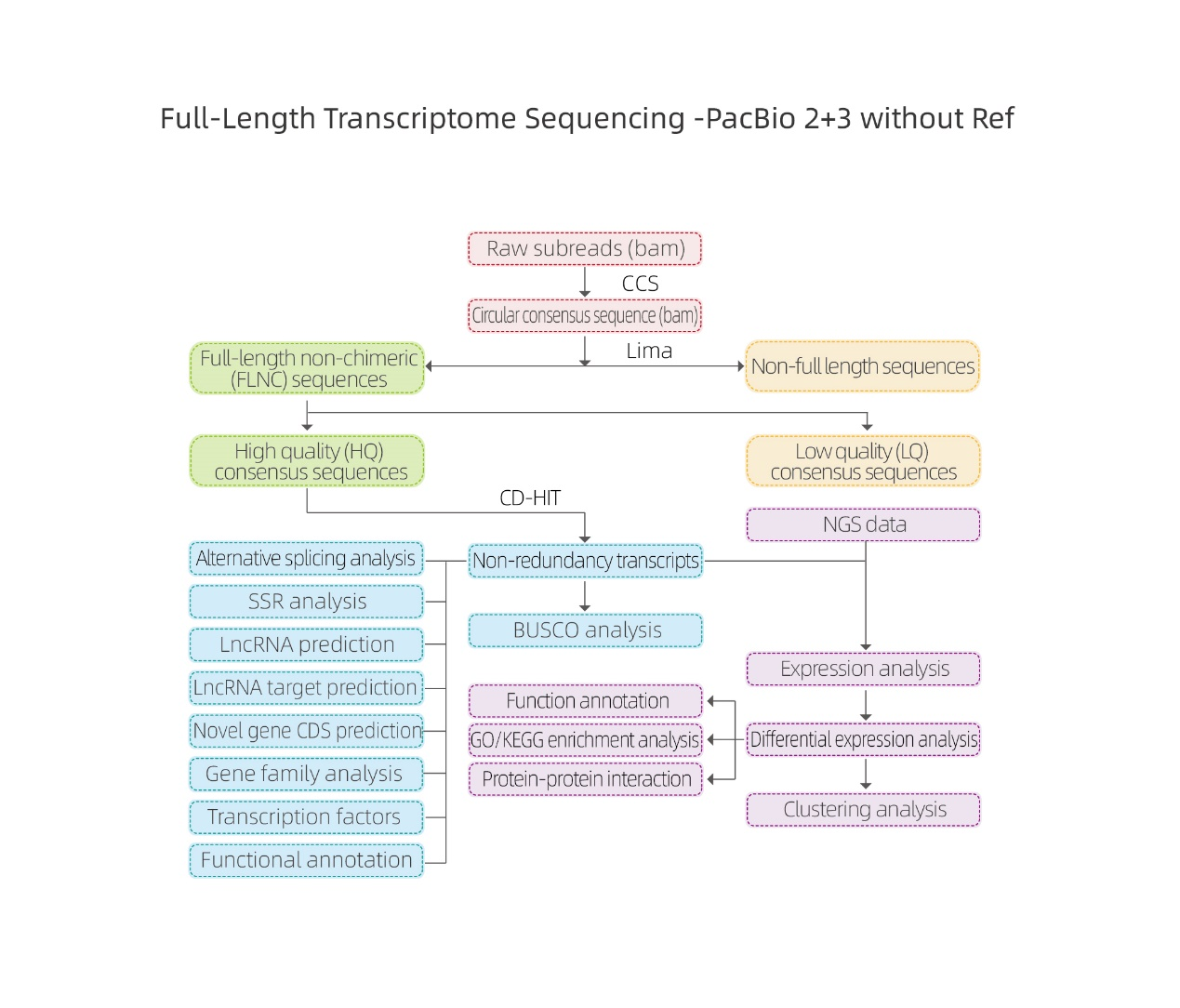
Análisis bioinformáticos.
● Procesamiento de datos sin procesar
● Identificación de transcripción
● Estructura de secuencia
● Cuantificación de expresiones
● Anotación de función
Requisitos de muestra y entrega
Requisitos de muestra:
Nucleótidos:
| Conc.(ng/μl) | Cantidad (μg) | Pureza | Integridad |
| ≥ 120 | ≥ 0,6 | DO260/280=1,7-2,5 DO260/230=0,5-2,5 En el gel se muestra contaminación limitada o nula de proteínas o ADN. | Para plantas: RIN≥7,5; Para animales: RIN≥8,0; 5,0≥ 28S/18S≥1,0; elevación basal limitada o nula |
Tejido: Peso (seco):≥1 gramos
*Para tejido de menos de 5 mg, recomendamos enviar una muestra de tejido congelada instantáneamente (en nitrógeno líquido).
Suspensión celular:Recuento de células = 3×106- 1×107
*Recomendamos enviar lisado celular congelado.En caso de que la celda cuente menos de 5×105, se recomienda la congelación instantánea en nitrógeno líquido, que es preferible para la microextracción.
Muestras de sangre:Volumen≥1 ml
Microorganismo:Masa ≥ 1 g
Entrega de muestra recomendada
Envase:
Tubo de centrífuga de 2 ml (no se recomienda papel de aluminio)
Etiquetado de muestras: Grupo+réplica, por ejemplo, A1, A2, A3;B1, B2, B3... ...
Envío:
1. Hielo seco: las muestras deben empaquetarse en bolsas y enterrarse en hielo seco.
2. Tubos RNAstable: las muestras de ARN se pueden secar en un tubo de estabilización de ARN (por ejemplo, RNAstable®) y enviarse a temperatura ambiente.
Flujo de trabajo del servicio

Diseño de experimentos

Entrega de muestra

extracción de ARN

construcción de biblioteca

Secuenciación

Análisis de los datos

Servicios postventa
1.Distribución de longitud FLNC
La longitud de la lectura no quimérica completa (FLNC) indica la longitud del ADNc en la construcción de la biblioteca.La distribución de longitud del FLNC es un indicador crucial para evaluar la calidad de la construcción de la biblioteca.
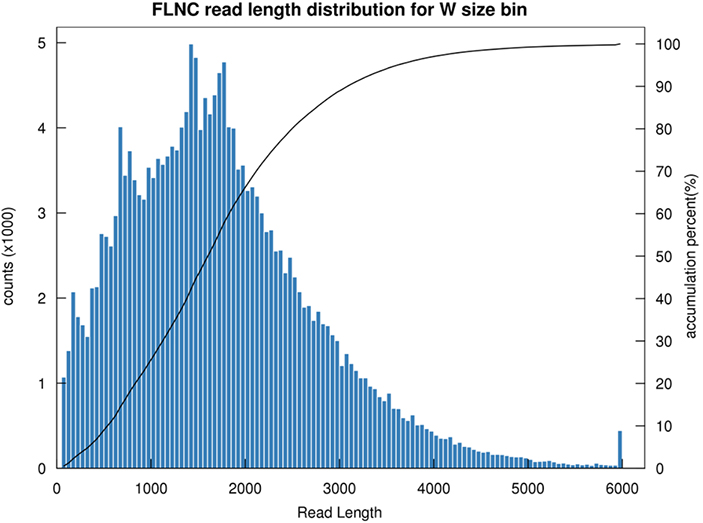
Distribución de longitud de lectura FLNC
2.Distribución completa de longitud de la región ORF
Utilizamos TransDecoder para predecir regiones codificantes de proteínas y las secuencias de aminoácidos correspondientes para generar conjuntos unigenes, que contienen información de transcripción completa no redundante en todas las muestras.
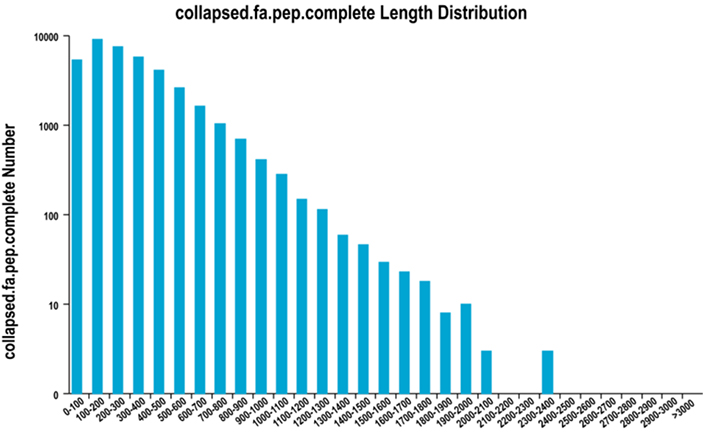
Distribución completa de longitud de la región ORF
3.Análisis de enriquecimiento de la vía KEGG
Las transcripciones expresadas diferencialmente (DET) se pueden identificar alineando los datos de secuenciación de ARN basados en NGS en conjuntos de transcripciones completos generados por los datos de secuenciación de PacBio.Estos DET pueden procesarse adicionalmente para diversos análisis funcionales, por ejemplo, análisis de enriquecimiento de la vía KEGG.
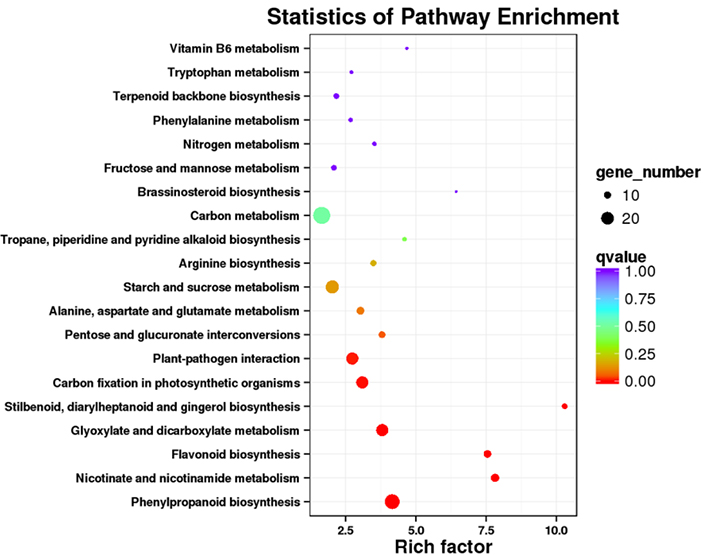
Enriquecimiento de la vía DET KEGG: diagrama de puntos
Caso BMK
La dinámica del desarrollo del transcriptoma de la raíz de Populus.
Publicado: Revista de biotecnología vegetal, 2019
Estrategia de secuenciación:
Coleccion de muestra:regiones del tallo: ápice, primer entrenudo (IN1), segundo entrenudo (IN2), tercer entrenudo (IN3), entrenudo (IN4) y entrenudo (IN5) de Nanlin895
Secuencia NGS:Se reunió el ARN de 15 individuos como una muestra biológica.Se procesaron tres réplicas biológicas de cada punto para la secuencia NGS.
Secuencia TGS:Las regiones del tallo se dividieron en tres regiones, es decir, ápice, IN1-IN3 e IN4-IN5.Cada región se procesó para la secuenciación PacBio con cuatro tipos de bibliotecas: 0-1 kb, 1-2 kb, 2-3 kb y 3-10 kb.
Resultados clave
1. Se identificaron un total de 87150 transcripciones completas, en las cuales se identificaron 2081 isoformas novedosas y 62058 isoformas empalmadas alternativas novedosas.
Se identificaron 2,1187 lncRNA y 356 genes de fusión.
3. Desde el crecimiento primario hasta el crecimiento secundario, se identificaron 15838 transcripciones expresadas diferencialmente de 995 genes expresados diferencialmente.En todos los DEG, 1216 eran factores de transcripción, la mayoría de los cuales aún no se han informado.
4.El análisis de enriquecimiento de GO reveló la importancia de la división celular y el proceso de oxidación-reducción en el crecimiento primario y secundario.
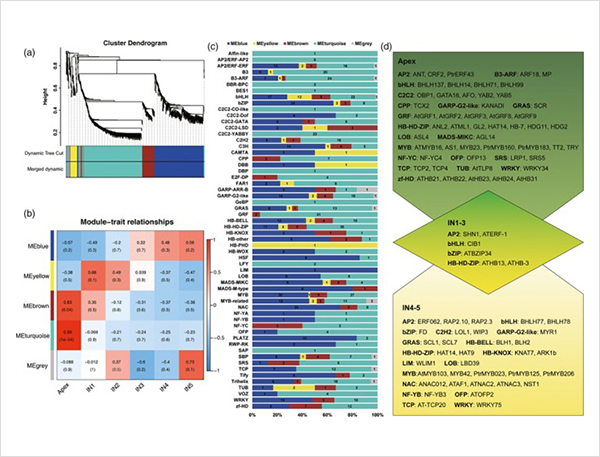
Eventos de empalme alternativos y diferentes isoformas.
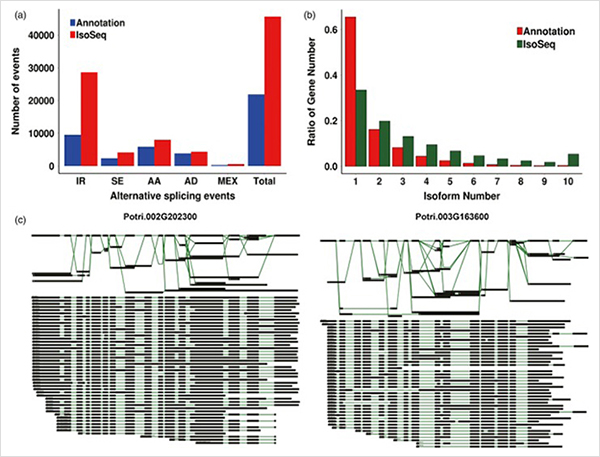
Análisis WGCNA sobre factores de transcripción.
Referencia
Chao Q, Gao ZF, Zhang D, et al.La dinámica del desarrollo del transcriptoma de la raíz de Populus.Plant Biotechnol J. 2019;17(1):206-219.doi:10.1111/pbi.12958





