

Spezifische Locus-amplifizierte Fragmentsequenzierung (SLAF-Seq)





Servicedetails
Technisches Schema
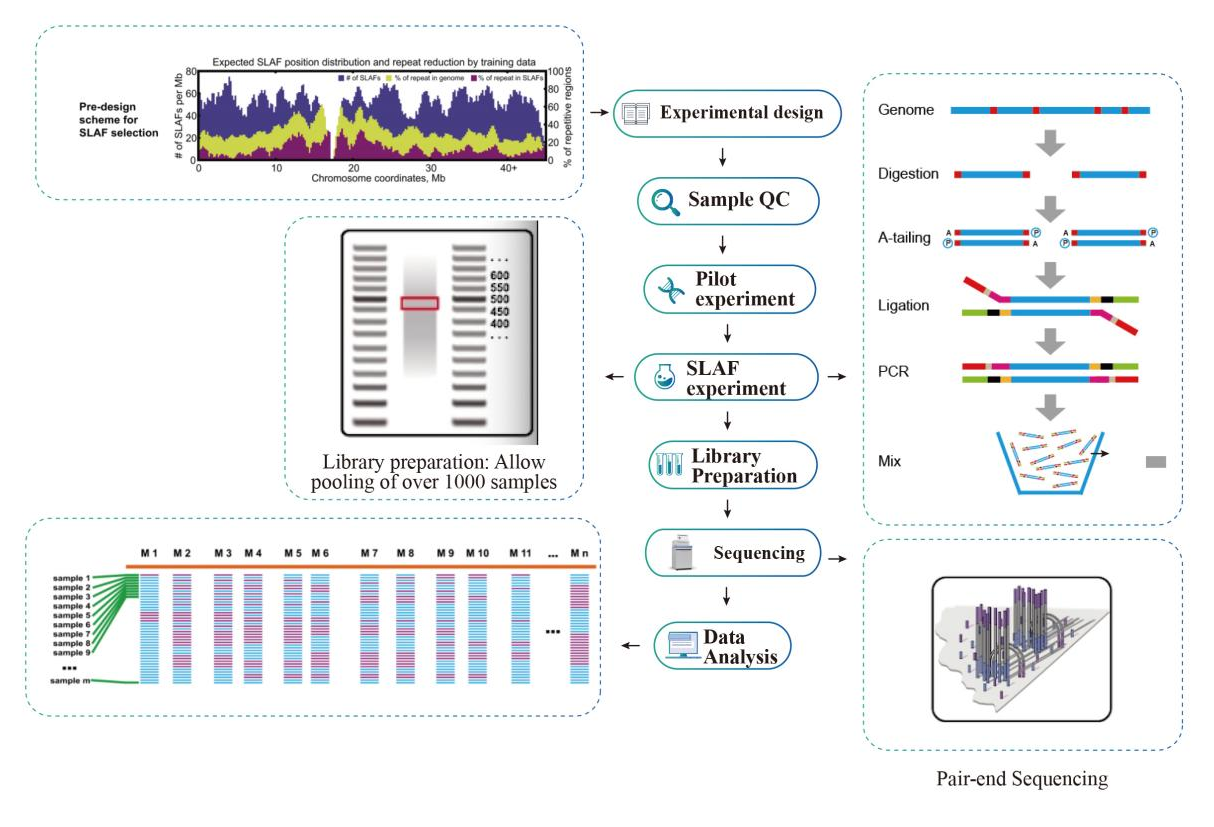
Arbeitsablauf

Servicevorteile
Hohe Effizienz bei der Markererkennung- Hochdurchsatz-Sequenzierungstechnologie unterstützt SLAF-Seq bei der Entdeckung Hunderttausender Tags im gesamten Genom.
Geringe Abhängigkeit vom Genom- Es kann auf Arten mit oder ohne Referenzgenom angewendet werden.
Flexible Schemagestaltung- Einzel-Enzym-, Dual-Enzym-, Multi-Enzym-Verdauung und verschiedene Arten von Enzymen können alle ausgewählt werden, um unterschiedliche Forschungsziele oder Spezies zu erfüllen.Eine Vorabbewertung in silico wird verwendet, um ein optimales Enzymdesign sicherzustellen.
Effiziente enzymatische Verdauung- Zur Optimierung der Bedingungen wurde ein Vorversuch durchgeführt, der das formale Experiment stabil und zuverlässig macht.Die Effizienz der Fragmentsammlung kann über 95 % erreichen.
Gleichmäßig verteilte SLAF-Tags- SLAF-Tags sind weitestgehend gleichmäßig in allen Chromosomen verteilt und erreichen durchschnittlich 1 SLAF pro 4 kb.
Effektive Vermeidung von Wiederholungen- Die Wiederholungssequenz in SLAF-Seq-Daten wird auf weniger als 5 % reduziert, insbesondere bei Arten mit einem hohen Anteil an Wiederholungen, wie Weizen, Mais usw.
Langjährige Erfahrung-Über 2000 abgeschlossene SLAF-Seq-Projekte zu Hunderten von Arten, darunter Pflanzen, Säugetiere, Vögel, Insekten, Wasserorganismen usw.
Selbst entwickelter bioinformatischer Workflow- Ein integrierter bioinformatischer Workflow für SLAF-Seq wurde von BMKGENE entwickelt, um die Zuverlässigkeit und Genauigkeit der Endausgabe sicherzustellen.
Leistungsbeschreibung
| Plattform | Konz. (ng/gl) | Gesamt (ug) | OD260/280 |
| Illumina NovaSeq | >35 | >1.6(Band>15μl) | 1,6-2,5 |
Empfohlene Sequenzierungsstrategie
Sequenzierungstiefe: 10X/Tag
| Genomgröße | Empfohlene SLAF-Tags |
| < 500 MB | 100K oder WGS |
| 500 MB – 1 GB | 100 K |
| 1 GB -2 GB | 200 K |
| Riesige oder komplexe Genome | 300 - 400K |
| Anwendungen
| Empfohlen Bevölkerungsskala
| Sequenzierungsstrategie und -tiefe
| |
| Tiefe
| Tag-Nummer
| ||
| GWAS
| Probenanzahl ≥ 200
| 10X
|
Entsprechend Genomgröße
|
| Genetische Evolution
| Einzelpersonen von jedem Untergruppe ≥ 10; Gesamtproben ≥ 30
| 10X
| |
Empfohlene Musterlieferung
Behälter: 2 ml Zentrifugenröhrchen
Für die meisten Proben empfehlen wir, sie nicht in Ethanol aufzubewahren.
Probenkennzeichnung: Proben müssen deutlich gekennzeichnet sein und mit dem eingereichten Probeninformationsformular identisch sein.
Versand: Trockeneis: Die Proben müssen zunächst in Beutel verpackt und in Trockeneis vergraben werden.
Service-Workflow






Proben-QC
Pilotversuch
SLAF-Experiment
Bibliotheksvorbereitung
Sequenzierung
Datenanalyse
Kundendienst
1. Statistik des Kartenergebnisses

2. Entwicklung des SLAF-Markers

3. Variationsanmerkung

| Jahr | Tagebuch | IF | Titel | Anwendungen |
| 2022 | Naturkommunikation | 17.694 | Genomische Basis der Giga-Chromosomen und des Giga-Genoms der Strauchpfingstrose Paeonia ostii | SLAF-GWAS |
| 2015 | Neuer Phytologe | 7.433 | Domestizierungs-Fußabdrücke verankern genomische Regionen von agronomischer Bedeutung in Sojabohnen | SLAF-GWAS |
| 2022 | Zeitschrift für fortgeschrittene Forschung | 12.822 | Genomweite künstliche Introgressionen von Gossypium barbadense in G. hirsutum zeigen überlegene Standorte für eine gleichzeitige Verbesserung der Baumwollfaserqualität und des Ertrags Züge | SLAF-Evolutionäre Genetik |
| 2019 | Molekulare Pflanze | 10.81 | Populationsgenomanalyse und De-Novo-Assemblierung enthüllen den Ursprung von Weedy Reis als evolutionäres Spiel | SLAF-Evolutionäre Genetik |
| 2019 | Naturgenetik | 31.616 | Genomsequenz und genetische Vielfalt des Karpfens Cyprinus carpio | SLAF-Linkage-Karte |
| 2014 | Naturgenetik | 25.455 | Das Genom kultivierter Erdnüsse bietet Einblick in die Karyotypen polyploider Hülsenfrüchte Evolution und Domestizierung von Nutzpflanzen. | SLAF-Linkage-Karte |
| 2022 | Zeitschrift für Pflanzenbiotechnologie | 9.803 | Die Identifizierung von ST1 offenbart eine Selektion, bei der die Samenmorphologie per Anhalter verändert wird und Ölgehalt während der Domestizierung von Sojabohnen | SLAF-Marker-Entwicklung |
| 2022 | Internationale Zeitschrift für Molekularwissenschaften | 6.208 | Identifizierung und DNA-Marker-Entwicklung für einen Weizen-Leymus mollis 2Ns (2D) Disomische Chromosomensubstitution | SLAF-Marker-Entwicklung |





