

GWAS
Titel: Die Neusequenzierung des gesamten Genoms enthüllt den Ursprung von Brassica napus und die genetischen Loci, die an seiner Verbesserung beteiligt sind
Tagebuch: Naturkommunikation
NGS |WGS |Neusequenzierung |GWAS |Transkriptom |RNAseq |Brassica napus |Entwicklung |Domestizierung
In dieser Studie stellte Biomarker Technologies Dienstleistungen zur NGS-Sequenzierung sowie technische Unterstützung bei der bioinformatischen Analyse von Sequenzierungsdaten bereit.
Hintergrund
Brassica napus(Raps) ist eine wichtige Ölsaatenpflanze und ein hervorragendes Modell für die Untersuchung von Prozessen der polyploiden Artbildung, Evolution und Selektion.Es ist jedoch noch unklar, ob Wildarten oder domestizierte Spender elterliche Vorfahren und Gene waren, die zur Domestizierung und Verbesserung des Rapses beitrugen.
Materialen und Methoden
Materialien:588B. napusAn dieser Studie waren Akzessionen beteiligt, darunter 466 aus Asien, 102 aus Europa, 13 aus Nordamerika und 7 aus Australien.Basierend auf Aufzeichnungen über die Wachstumsgewohnheiten wurden diese Materialien in drei Ökotypen unterteilt;Frühling (86 Beitritte), Winter (74 Beitritte) und Halbwinter (428 Beitritte).
Reihenfolge:Durchschnittlich ca.5× (im Bereich von 3,37× bis 7,71×)
Sequenzierungsplattform:Illumina Hiseq 4000
Datenproduktion:4,03 TB saubere Daten
SNP-Aufruf:BWA + GATK.Es wurden 5.294.158 SNPs und 1.307.151 InDels erhalten.
Ergebnisse
Herkunft von B. napus
B. napusAus dem Vorfahren der europäischen Rübe entwickelte sich ein Subgenom.Ein Genflussereignis von der europäischen Rübe zurB. napus Ein Subgenom entstand vor etwa 106–1170 Jahren.B. napusDas C-Subgenom könnte sich aus dem gemeinsamen Vorfahren dieser Abstammungslinien entwickelt haben.Der Vorfahre vonB. napusAbspaltung vom gemeinsamen Vorfahren von vier B. oleracea-Unterarten, in die kürzlich ein Gen eingeflossen istB. napusVor ca. 108–898 Jahren.B. napusDas C-Subgenom hat einen komplexeren Ursprung als das A-Subgenom.Dabei entwickelte sich in beiden Subgenomen ein starker EngpassB. napusEvolution.Der Winter und HalbwinterB. napusDie Ökotypen unterschieden sich vor etwa 60 Jahren, während Winter und Frühling unterschieden wurdenB. napusvor ca. 416 Jahren auseinander gingen, und Ölsaaten und Nicht-ÖlsaatenB. napusdivergierte vor ca. 277 Jahren.
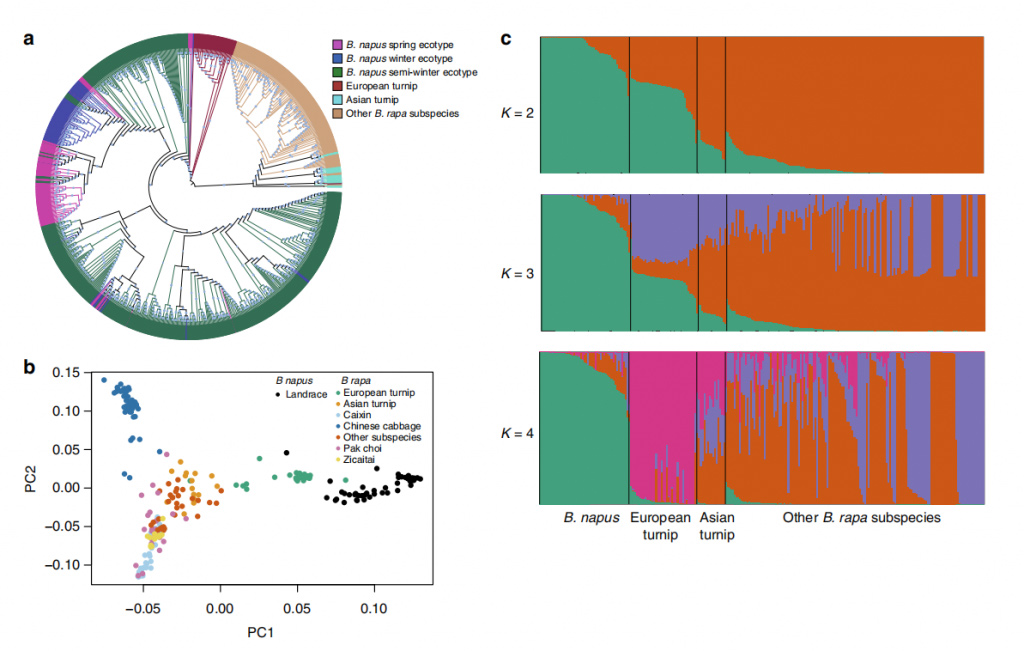
Abb. 2 Populationsstruktur von 588 B. napus-Akzessionen und 199 B. rapa-Akzessionen.
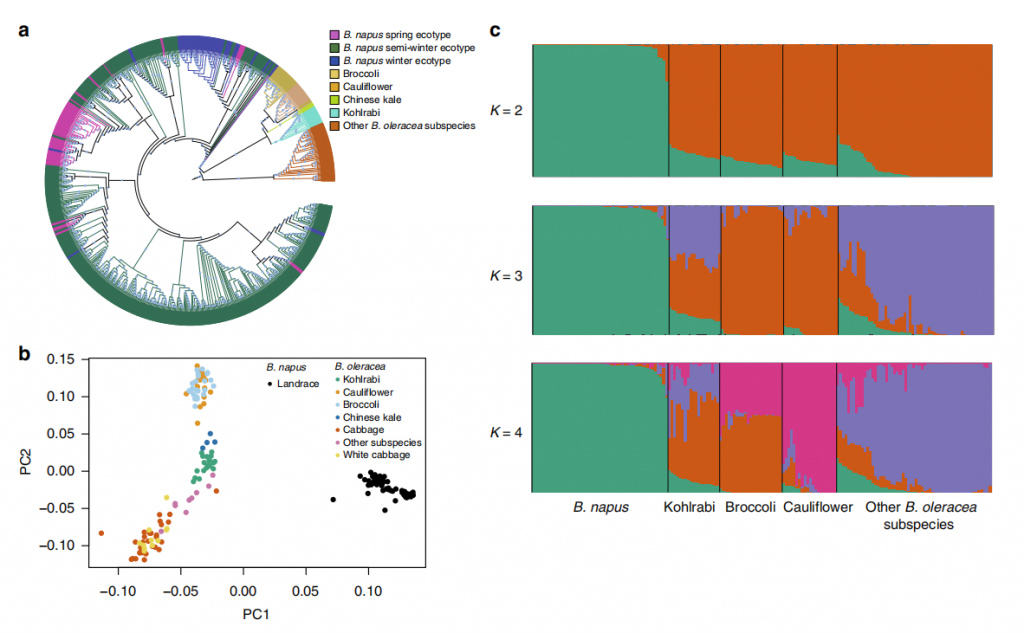
Abb. 3 Populationsstruktur von 588 B. napus-Akzessionen und 119 B. oleracea-Akzessionen
Selektionssignale und genomweite Assoziationsstudien.
Während der ersten Verbesserungsphase (FSI) ging im C-Subgenom von B. napus mehr genetische Vielfalt verloren als im A-Subgenom.Während des FSI kam es zu einer geringeren genetischen Differenzierung als während der zweiten Verbesserungsphase (SSI).Gene in SSI-Selektionssignalregionen wurden hinsichtlich Stresstoleranz, Entwicklung und Stoffwechselwegen angereichert.Es wurden 60 Loci identifiziert, die signifikant mit 10 Zielmerkmalen assoziiert sind, darunter 5 im Zusammenhang mit dem Samenertrag, 3 mit der Schotenlänge, 4 mit dem Ölgehalt und 48 mit der Samenqualität.
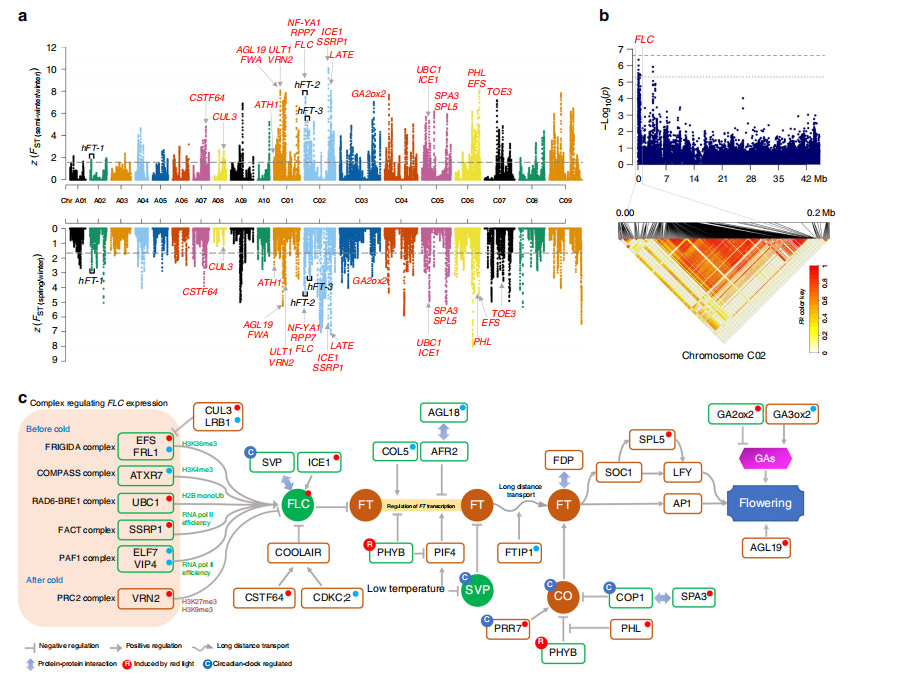
Abb. 4 Genomweites Scannen und Annotationen ausgewählter Regionen während der SSI von B. napus
Transkriptomanalyse
RNAseq-Daten von 11 Geweben einer Sorte mit hohem Ölgehalt und doppelt niedrigem Ölgehalt sowie einer Sorte mit niedrigem Ölgehalt und doppelt hohem Ölgehalt identifizierten Gene, die mit dem Glucosinolat-Biosyntheseprozess in Zusammenhang stehen, waren deutlich überrepräsentiert.
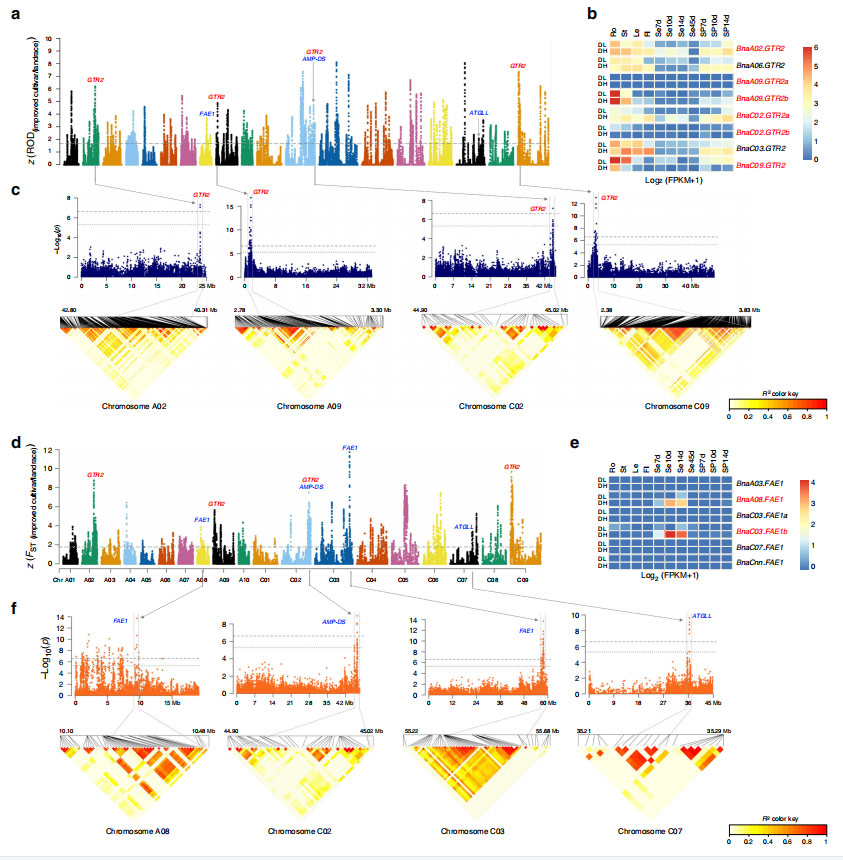
Abb. 5 Übersicht über die Blühzeitregulierung unter Auswahl der Ökotypverbesserung von B. napus
Diskussion
Diese Studie lieferte eine wertvolle Ressource zum Verständnis des Ursprungs und der Verbesserungsgeschichte vonB. napusund wird die Analyse der genetischen Grundlagen wichtiger agronomischer Komplexmerkmale erleichtern.Die bedeutenden SNPs, die mit günstigen Varianten, Selektionssignalen und Kandidatengenen verbunden sind, werden in Zukunft einen großen Beitrag leisten, insbesondere zur Verbesserung des Ertrags, der Samenqualität, des Ölgehalts und der Anpassungsfähigkeit dieser neuen allopolyploiden Kulturpflanze und ihrer Verwandten.
Referenz
Die Neusequenzierung des gesamten Genoms enthüllt den Ursprung von Brassica napus und die genetischen Loci, die an seiner Verbesserung beteiligt sind[J].Naturkommunikation, 2019, 10(1).
Neuigkeiten und Highlights Ziel ist es, die neuesten erfolgreichen Fälle mit Biomarker-Technologien zu teilen und neue wissenschaftliche Errungenschaften sowie herausragende Techniken, die während der Studie angewendet wurden, zu erfassen.
Zeitpunkt der Veröffentlichung: 05.01.2022