

Die Hochdurchsatz-Genotypisierung, insbesondere bei großen Populationen, ist ein grundlegender Schritt in genetischen Assoziationsstudien, der die genetische Grundlage für die Entdeckung funktioneller Gene, die Evolutionsanalyse usw. liefert. Anstelle der tiefgreifenden Neusequenzierung des gesamten Genoms wird die Genomsequenzierung mit reduzierter Repräsentation (RRGS) verwendet ) wird eingeführt, um die Sequenzierungskosten pro Probe zu minimieren und gleichzeitig eine angemessene Effizienz bei der Entdeckung genetischer Marker aufrechtzuerhalten.Dies wird üblicherweise durch Extrahieren von Restriktionsfragmenten innerhalb eines bestimmten Größenbereichs erreicht, der als „Reduced Representation Library“ (RRL) bezeichnet wird.Specific-Locus Amplified Fragment Sequencing (SLAF-Seq) ist eine selbst entwickelte Strategie für die De-novo-SNP-Entdeckung und SNP-Genotypisierung großer Populationen.
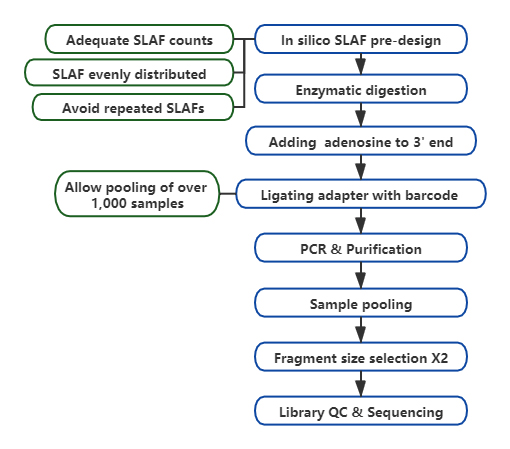
Technischer Arbeitsablauf
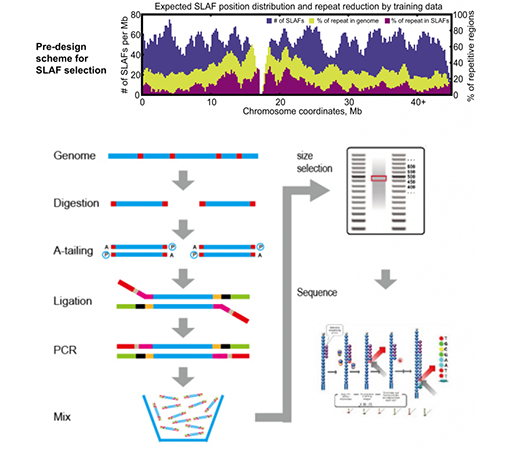
SLAF im Vergleich zu bestehenden RRL-Methoden
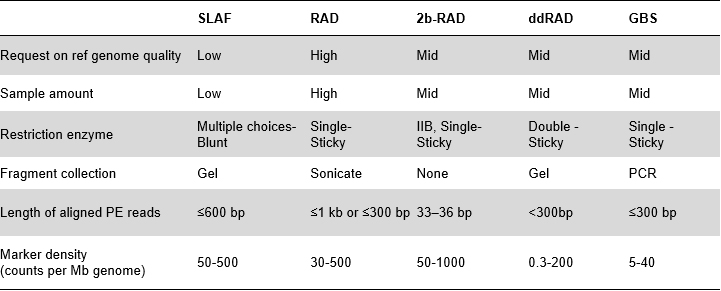
Vorteile von SLAF
Höhere Effizienz bei der Entdeckung genetischer Marker– In Kombination mit der Hochdurchsatz-Sequenzierungstechnologie könnte SLAF-Seq Hunderttausende von Tags im gesamten Genom entdecken, um die Anforderungen verschiedener Forschungsprojekte zu erfüllen, entweder mit oder ohne Referenzgenom.
Maßgeschneidertes und flexibles experimentelles Design– Für unterschiedliche Forschungsziele oder Spezies stehen verschiedene enzymatische Verdauungsstrategien zur Verfügung, darunter Einzelenzym-, Doppelenzym- und Multienzym-Verdauung.Die Verdauungsstrategie wird vorab in silico evaluiert, um ein optimales Enzymdesign sicherzustellen.
Hohe Effizienz bei der enzymatischen Verdauung– Die vorgefertigte enzymatische Verdauung sorgt für eine gleichmäßigere Verteilung der SLAFs auf dem Chromosom.Die Effizienz der Fragmentsammlung kann über 95 % erreichen.
Vermeiden Sie sich wiederholende Abläufe– Der Prozentsatz der repetitiven Sequenz in den SLAF-Seq-Daten wird auf weniger als 5 % reduziert, insbesondere bei Arten mit einem hohen Anteil an repetitiven Elementen, wie Weizen, Mais usw.
Selbst entwickelter bioinformatischer Workflow– BMK hat einen integrierten bioinformatischen Workflow entwickelt, der auf die SLAF-Seq-Technologie anwendbar ist, um Zuverlässigkeit und Genauigkeit der Endausgabe sicherzustellen.
Anwendung von SLAF
Genetische Verknüpfungskarte
Erstellung einer genetischen Karte mit hoher Dichte und Identifizierung von Loci, die Blütentypmerkmale in Chrysanthemen (Chrysanthemum x morifolium Ramat) steuern.
Zeitschrift: Horticulture Research Veröffentlicht: 2020.7

GWAS
Identifizierung eines Kandidatengens, das mit dem Isofavongehalt in Sojabohnensamen assoziiert ist, mittels genomweiter Assoziations- und Verknüpfungskartierung
Zeitschrift: The Plant Journal Veröffentlicht: 2020.08

Evolutionäre Genetik
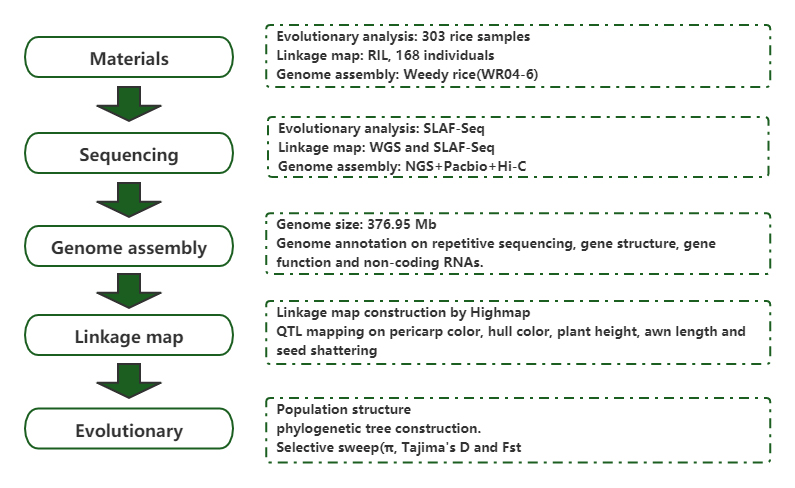
Populationsgenomanalysen und De-novo-Zusammensetzung offenbaren den Ursprung von Unkrautreis als evolutionäres Spiel
Zeitschrift: Molecular Plant Veröffentlicht: 2019.5
Bulked Segregant Analysis (BSA)
GmST1, das eine Sulfotransferase kodiert, verleiht Resistenz gegen die Sojabohnenmosaikvirusstämme G2 und G3
Zeitschrift: Plant, Cell&Environment Veröffentlicht: 2021.04

Referenz
Sun X, Liu D, Zhang X, et al.SLAF-Seq: eine effiziente Methode zur groß angelegten De-novo-SNP-Entdeckung und Genotypisierung mittels Hochdurchsatzsequenzierung[J].Plus eins, 2013, 8(3):e58700
Song X, Xu Y, Gao K, et al.Erstellung einer genetischen Karte mit hoher Dichte und Identifizierung von Loci, die Blütentypmerkmale in Chrysanthemen (Chrysanthemum × morifolium Ramat.) steuern.Hortic Res.2020;7:108.
Wu D, Li D, Zhao X, et al.Identifizierung eines Kandidatengens, das mit dem Isoflavongehalt in Sojabohnensamen assoziiert ist, mittels genomweiter Assoziations- und Verknüpfungskartierung.Werk J. 2020;104(4): 950-963.
Sun J, Ma D, Tang L, et al.Populationsgenomanalyse und De-Novo-Assemblierung enthüllen den Ursprung von Weedy Rice als evolutionäres Spiel.Mol-Pflanze.2019;12(5):632-647.Mol-Pflanze.2018;11(11):1360-1376.
Zhao X, Jing Y, Luo Z, et al.GmST1, das eine Sulfotransferase kodiert, verleiht Resistenz gegen die Sojabohnenmosaikvirusstämme G2 und G3.Pflanzenzellumgebung.2021;10.1111/Stk.14066
Zeitpunkt der Veröffentlichung: 04.01.2022