

Transkriptomik
Natur
KOMMUNIKATION
Die vollständige Transkriptcharakterisierung der SF3B1-Mutation bei chronischer lymphatischer Leukämie zeigt eine Herunterregulierung zurückgehaltener Introns
Transkripte in voller Länge|Nanoporensequenzierung|Alternative Isoformenanalyse
Hintergrund
SEs wurde weithin berichtet, dass omatische Mutationen im Spleißfaktor SF3B1 mit verschiedenen Krebsarten in Zusammenhang stehen, darunter chronische lymphatische Leukämie (CLL), Aderhautmelanom, Brustkrebs usw. Darüber hinaus haben kurze transkriptomische Studien abweichende Spleißmuster aufgedeckt, die durch SF3B1-Mutationen hervorgerufen werden.Studien zu diesen alternativen Spleißmustern beschränkten sich jedoch lange Zeit auf die Ereignisebene und es mangelte an Wissen über die Isoformenebene, da nur kurze lesbare zusammengestellte Transkripte zur Verfügung standen.Hier wurde eine Nanoporen-Sequenzierungsplattform eingeführt, um Transkripte in voller Länge zu generieren, die die Untersuchung von AS-Isoformen ermöglichte.
Experimentelles Design
Experimente
Gruppierung:1. CLL-SF3B1(WT) 2. CLL-SF3B1(K700E-Mutation);3. Normale B-Zellen
Sequenzierungsstrategie:MinION 2D-Bibliothekssequenzierung, PromethION 1D-Bibliothekssequenzierung;Kurzlesen von Daten aus denselben Proben
Sequenzierungsplattform:ONT MiniON;ONT PromethION;
Bioinformatische Analyse
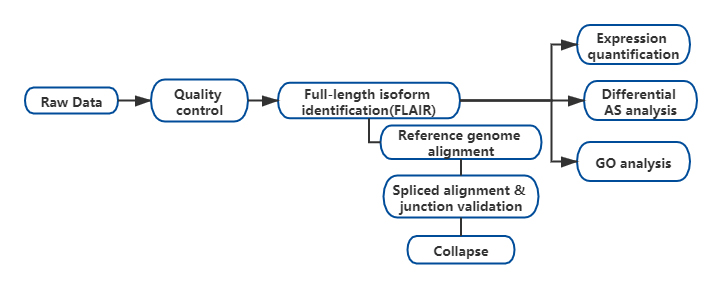
Ergebnisse
AInsgesamt wurden 257 Millionen Lesevorgänge aus 6 CLL-Proben und 3 B-Zellen generiert.Im Durchschnitt wurden 30,5 % dieser Lesevorgänge als Transkripte in voller Länge identifiziert.
FDie Analyse alternativer RNA-Isoformen in voller Länge (FLAIR) wurde entwickelt, um eine Reihe hochzuverlässiger Isoformen zu generieren.FLAIR kann wie folgt zusammengefasst werden:
NAnopore liest Alignment: Identifiziert die allgemeine Transkriptstruktur basierend auf dem Referenzgenom;
SPlice-Junction-Korrektur: Korrigieren Sie Sequenzfehler (rot) mit der Spleißstelle von entweder annotierten Introns, Introns aus Short-Read-Daten oder beiden;
COllapse: Fassen Sie repräsentative Isoformen basierend auf Spleißverbindungsketten zusammen (First-Pass-Set).Wählen Sie ein ISOFrom mit hoher Zuverlässigkeit basierend auf der Anzahl der unterstützenden Lesevorgänge aus (Schwellenwert: 3).
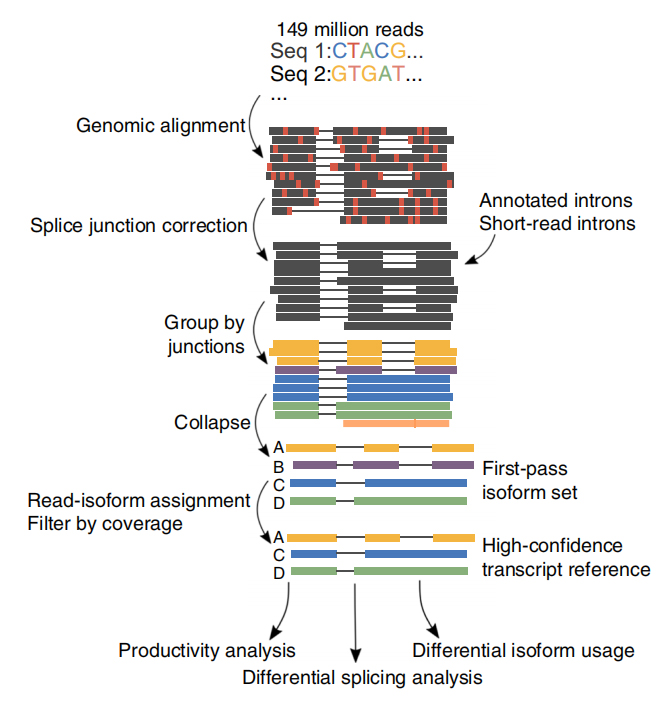
Abbildung 1. FLAIR-Analyse zur Identifizierung vollständiger Isoformen, die mit der SF3B1-Mutation bei CLL assoziiert sind
FLAIR identifizierte 326.699 hochzuverlässige gespleißte Isoformen, von denen 90 % neuartige Isoformen sind.Bei den meisten dieser nicht annotierten Isoformen handelte es sich um neuartige Kombinationen bekannter Spleißverbindungen (142.971), während die übrigen neuen Isoformen entweder beibehaltenes Intron (21.700) oder neues Exon (3594) enthielten.
LOng-Read-Sequenzen ermöglichen die Identifizierung mutierter SF3B1-K700E-veränderter Spleißstellen auf Isoformenebene.Es wurde festgestellt, dass 35 alternative 3'SSs und 10 alternative 5'SSs zwischen SF3B1-K700E und SF3B1-WT signifikant unterschiedlich gespleißt waren.33 der 35 Veränderungen wurden durch lange Lesesequenzen neu entdeckt.In Nanopore-Daten beträgt die Abstandsverteilung zwischen SF3B1-K700E-veränderten 3'SSs zu kanonischen Standortspitzen etwa -20 bp, was sich deutlich von einer Kontrollverteilung unterscheidet, ähnlich dem, was in CLL-Short-Read-Sequenzen berichtet wurde.Isoformen des ERGIC3-Gens wurden analysiert, wobei eine neue Isoform, die die proximale Spleißstelle enthielt, häufiger in SF3B1-K700E gefunden wurde.Sowohl das proximale als auch das distale 3'SS waren mit ausgeprägten AS-Mustern verbunden, die mehrere Isoformen erzeugten.
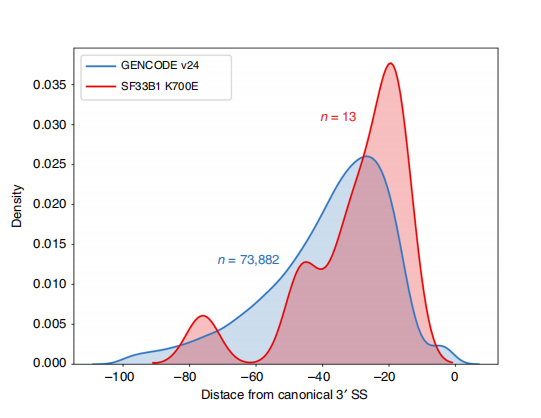
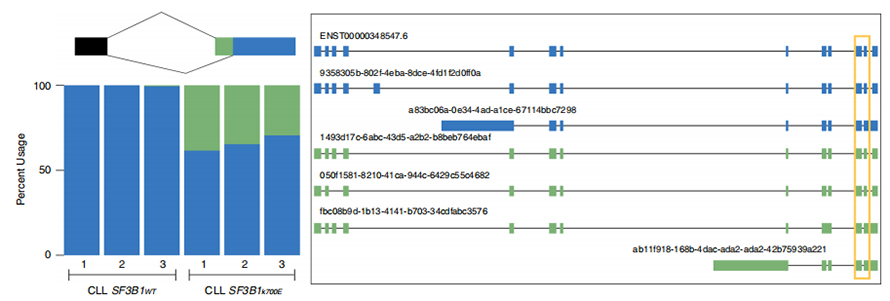
Abbildung 2. Alternative 3′-Spleißmuster, identifiziert mit Nanoporen-Sequenzierungsdaten
Aufgrund des Vertrauens in die IR-Identifizierung und -Quantifizierung war die Analyse der IR-Ereignisnutzung lange Zeit auf Short-Read-basierte Analysen beschränkt.Die Expression von IR-Isoformen in SF3B1-K700E und SF3B1-WT wurde anhand von Nanoporensequenzen quantifiziert, was eine globale Herunterregulierung der IR-Isoformen in SF3B1-K700E ergab.
Abbildung 4. Intensität der Landwirtschaft und Netzwerkkonnektivität zwischen drei Landwirtschaftssystemen (A und B);Zufällige Waldanalyse (C) und Zusammenhang zwischen landwirtschaftlicher Intensität und AMF-Besiedlung (D)
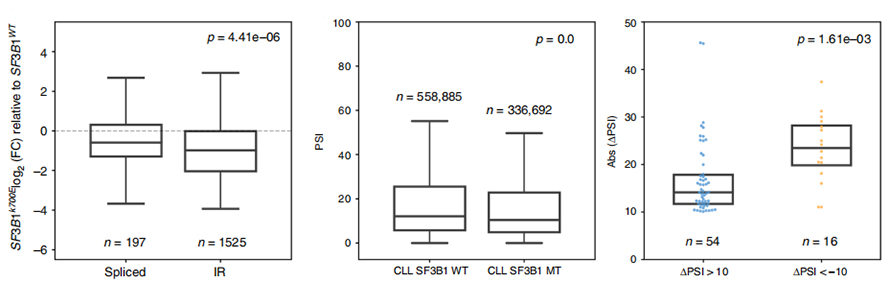
Abbildung 3. Intron-Retention-Ereignisse werden in CLL SF3B1-K700E stärker herunterreguliert
Technologie
Nanoporen-Long-Read-Sequenzierung
NAnopore-Sequenzierung ist eine Technologie zur Echtzeit-Sequenzierung elektrischer Signale einzelner Moleküle.
DDoppelsträngige DNA oder RNA binden an nanoporöses Protein, das im Biofilm eingebettet ist, und entfalten sich unter der Führung des Motorproteins.
DNA/RNA-Stränge passieren das Nanoporen-Kanalprotein unter der Wirkung von Spannungsunterschieden mit einer bestimmten Geschwindigkeit.
MMoleküle erzeugen je nach chemischer Struktur unterschiedliche elektrische Signale.
RDie Echtzeiterkennung von Sequenzen wird durch Base Calling erreicht.
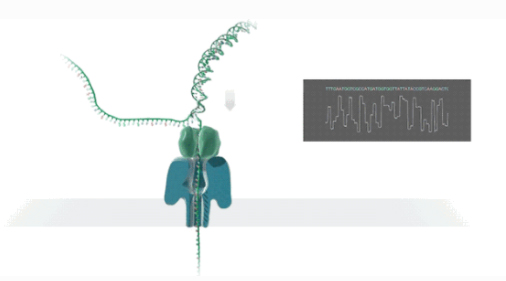
Durchführung einer Transkriptomsequenzierung in voller Länge
√ Datensättigung
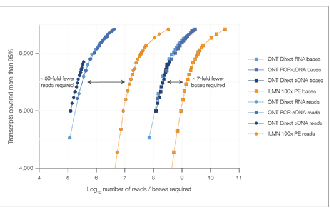
Es sind 7-mal weniger Lesevorgänge erforderlich, um eine vergleichbare Datensättigung zu erreichen.
√ Identifizierung der Transkriptstruktur
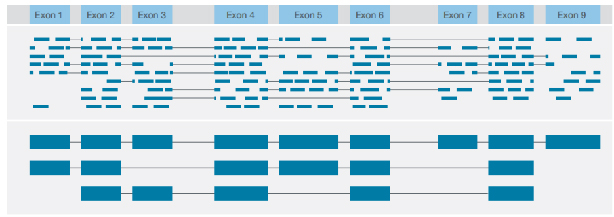
Identifizierung verschiedener Strukturvarianten mit Konsensauslesung jedes Transkripts in voller Länge
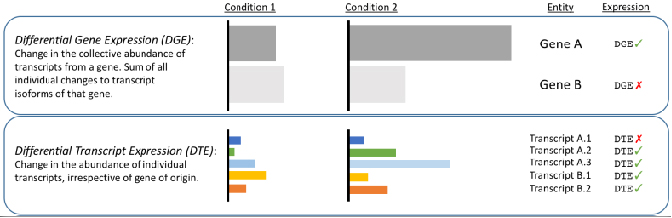
√ Differentialanalyse auf Transkriptebene – Durch kurze Lesevorgänge verborgene Änderungen aufdecken
Referenz
Tang AD, Soulette CM, Baren MJV, et al.Die vollständige Transkriptcharakterisierung der SF3B1-Mutation bei chronischer lymphatischer Leukämie zeigt eine Herunterregulierung zurückgehaltener Introns[J].Naturkommunikation.
Technik und Highlights Ziel ist es, die neuesten erfolgreichen Anwendungen verschiedener Hochdurchsatz-Sequenzierungstechnologien in verschiedenen Forschungsbereichen sowie brillante Ideen im experimentellen Design und beim Data Mining zu teilen.
Zeitpunkt der Veröffentlichung: 08.01.2022