

GENOM-EVOLUTION
Naturgenetik
Eine qualitativ hochwertige Genomassemblierung hebt die genomischen Merkmale und agronomisch wichtigen Gene des Roggens hervor
PacBio |Illumina |Optische Karte von Bionano |Hi-C-Genomassemblierung |Genetische Karte |Selektive Sweeps |RNA-Seq |ISO-seq |SLAF-seq
Biomarker Technologies leistete in dieser Studie technische Unterstützung bei der Pacbio-Sequenzierung, Hi-C-Sequenzierung und Datenanalyse.
Höhepunkte
1. Es wurde das erste hochwertige Roggengenom auf Chromosomenebene erhalten, das eine einzelne Chromosomengröße von mehr als 1 GB aufweist.
2. Im Vergleich zum Tu-, Aet- und Hv-Genom wurde kürzlich ein einzigartiges LTR-RT-Ereignis im Roggengenom beobachtet, das für die Vergrößerung der Roggengenomgröße verantwortlich war.
3. Die Divergenz zwischen Roggen und diploiden Weizen erfolgte nach der Trennung von Gerste und Weizen, wobei die Divergenzzeiten für die beiden Ereignisse etwa 9,6 und 15 MYA betrugen.
Die Phosphorylierung von FT-Genen könnte das frühe Triebmerkmal bei Roggen steuern.
4. Selektive Sweep-Analysen deuten auf eine mögliche Beteiligung von ScID1 an der Regulierung des Erntedatums und seine wahrscheinliche Auswahl durch Domestizierung in Roggen hin
Hintergrund
Hintergrund
Roggen ist eine wertvolle Nahrungs- und Futterpflanze, eine wichtige genetische Ressource für die Verbesserung von Weizen und Triticale und ein unverzichtbares Material für effiziente vergleichende Genomstudien an Gräsern.Weining-Roggen, eine in China angebaute Frühblühersorte, zeichnet sich durch eine breite Resistenz gegen Echten Mehltau und Streifenrost aus.Um die genetischen und molekularen Grundlagen der Roggen-Elitemerkmale zu verstehen und die Genom- und Züchtungsstudien an Roggen und verwandten Nutzpflanzen voranzutreiben, haben wir hier das Genom von Weining-Roggen sequenziert und analysiert.
Erfolge
Roggengenom
Das Rye-Genom wurde durch Kombination von PacBio-SMRT-Reads, Short-Read-Illumina-Sequenzierung sowie solchen aus der Chromatin-Konformationserfassung (Hi-C), der genetischen Kartierung und der BioNano-Analyse konstruiert.Die zusammengesetzten Contigs (7,74 Gb) machten 98,47 % der geschätzten Genomgröße (7,86 Gb) aus, wobei 93,67 % der Contigs (7,25 Gb) sieben Chromosomen zugeordnet waren.Repetitive Elemente machten 90,31 % des zusammengesetzten Genoms aus.
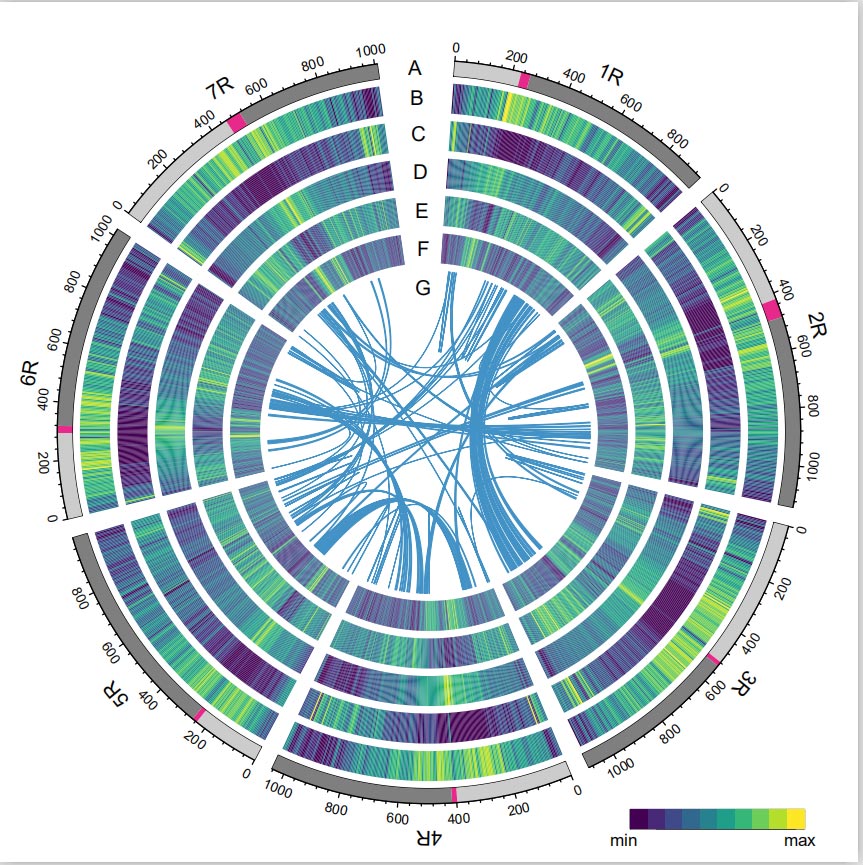
Roggengenom
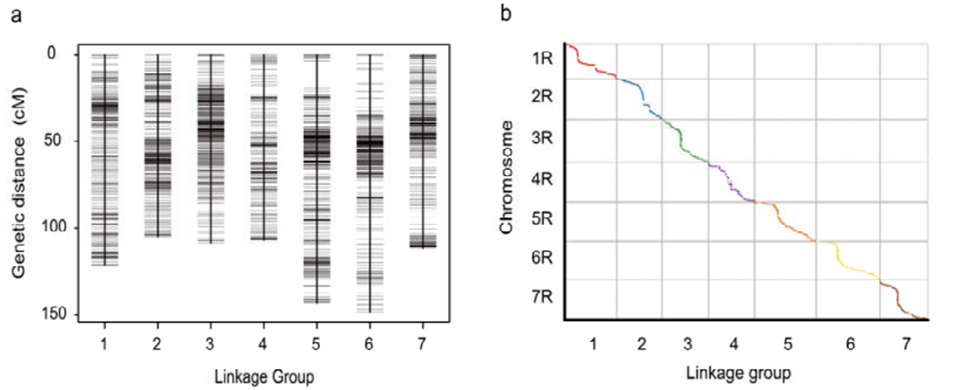
Genetische Verknüpfungskarte (WJ), entwickelt unter Verwendung von 295 F2-Pflanzen, die aus der Kreuzung zweier Roggenlandsorten (Weining × Jingzhou) stammen.
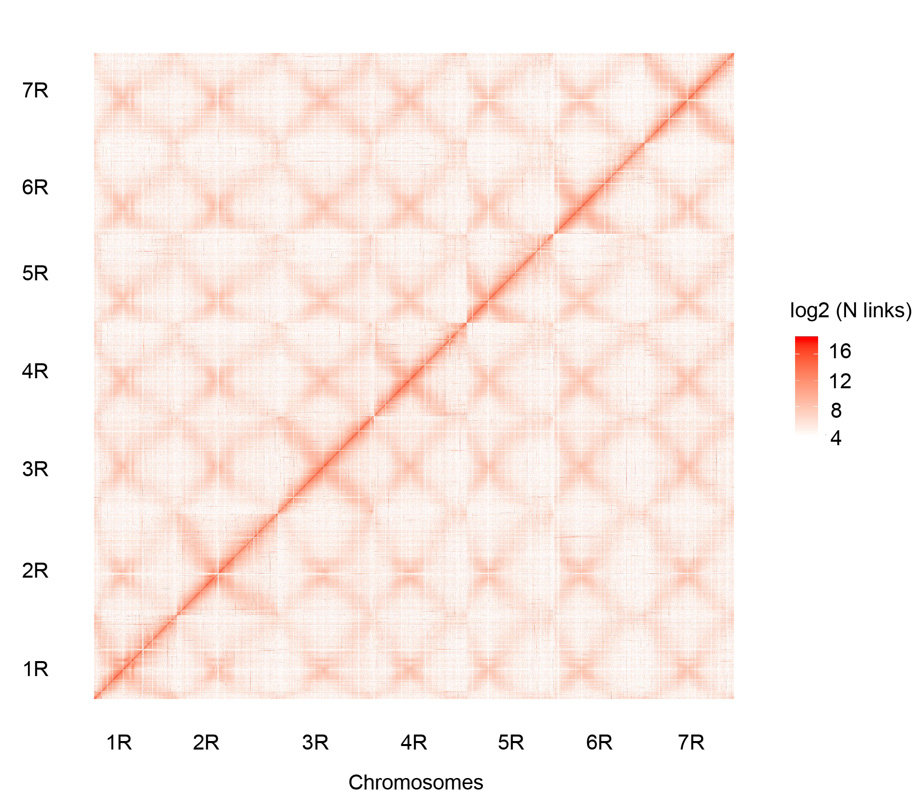
Hi-C-Kontaktkarte der sieben zusammengesetzten Weining-Roggenchromosomen (1R – 7R)
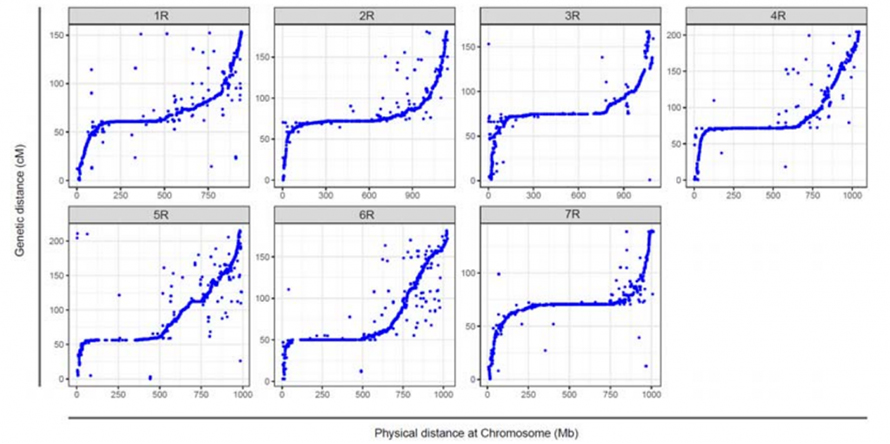
Ausrichtung zwischen den sieben zusammengesetzten Chromosomen von Weining-Roggen und den sieben Roggen-Verknüpfungsgruppen, die mithilfe der Lo7 x Lo255-RIL-Population entwickelt wurden
Der LTR Assembly Index (LAI)-Wert des Roggengenoms betrug 18,42 und 1.393 (96,74 %) der 1.440 hochkonservierten BUSCO-Gene wurden identifiziert. Diese Ergebnisse legen nahe, dass die Weining-Roggengenomsequenz in beiden intergenen Bereichen von hoher Qualität ist und Genregionen.Insgesamt wurden 86.991 proteinkodierende Gene vorhergesagt, darunter 45.596 Gene mit hohem Vertrauen (HC) und 41.395 Gene mit geringem Vertrauen (LC).
2. Analyse von TEs
Analyse von TEs.Insgesamt 6,99 GB, was 90,31 % der Weining-Assembly entspricht, wurden als TEs annotiert, darunter 2.671.941 Elemente, die zu 537 Familien gehörten.Dieser TE-Gehalt war deutlich höher als der zuvor für Ta (84,70 %), Tu (81,42 %), Aet (84,40 %), WEW (82,20 %) oder Hv (80,80 %) gemeldete.Die langen terminalen Wiederholungsretrotransposons (LTR-RTs), einschließlich Gypsy-, Copia- und nicht klassifizierter RT-Elemente, waren die dominanten TEs und machten 84,49 % des annotierten TE-Gehalts und 76,29 % des zusammengesetzten Weining-Genoms aus;CACTA-DNA-Transposons waren die zweithäufigsten TEs und machten 11,68 % des annotierten TE-Gehalts und 10,55 % des zusammengesetzten Weining-Genoms aus.
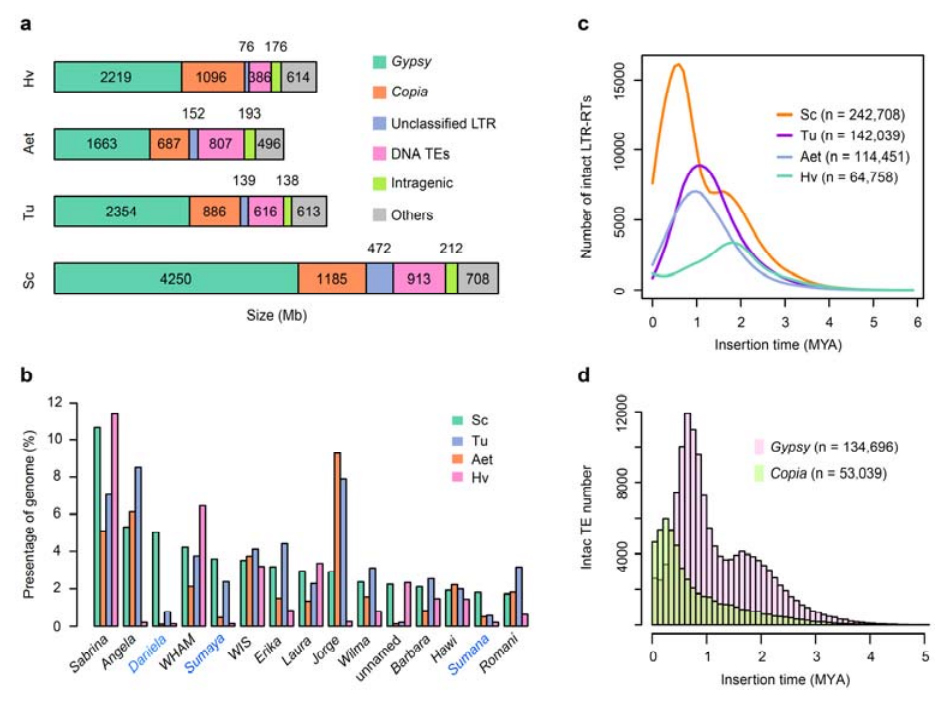
Analyse von Transposonelementen von Roggen
Weining-Roggen wies einen vergleichsweise hohen Anteil neuer LTR-RT-Insertionen auf, wobei der Höhepunkt der Amplifikation vor etwa 0,5 Millionen Jahren (MYA) auftrat, was der jüngste unter den vier Arten war;Der andere Peak trat vor etwa 1,7 MYA auf, war älter und wurde auch in Gerste beobachtet.Auf Superfamilienebene wurden sehr aktuelle Ausbrüche von Copia-Elementen in Weining-Roggen bei 0,3 MYA gefunden, während die Amplifikationen von Gypsy-RTs das bimodale Verteilungsmuster der LTR-RT-Burst-Dynamik maßgeblich prägten.
3. Untersuchung der Evolution des Roggengenoms und der Chromosomensyntenien
Die Divergenz zwischen Roggen und diploiden Weizen erfolgte nach der Trennung von Gerste und Weizen, wobei die Divergenzzeiten für die beiden Ereignisse etwa 9,6 bzw. 15 MYA betrugen.1R, 2R und 3R waren vollständig kollinear mit den Chromosomen der Gruppen 1, 2 und 3 von Weizen.4R, 5R, 6R, 7R wurden in großräumigen Fusionen und Segmenten gefunden.
4. Analyse von Genduplikationen und deren Einfluss auf Stärkebiosynthesegene
Bemerkenswerterweise war die Anzahl der tandemly duplizierten Gene (TDGs) und der proximal duplizierten Gene (PDGs) von Weining-Roggen höher als die für Tu, Aet, Hv, Bd und Os gefundenen.Transponierte duplizierte Gene (TrDGs) waren ebenfalls zahlreicher als diejenigen, die speziell für Tu und Aet gefunden wurden.Die Erweiterung des Roggengenoms geht mit einer höheren Anzahl an Genduplikationen einher.Die erhöhten TE-Ausbrüche im Roggen könnten zu einer erhöhten Anzahl von TrDGs geführt haben.
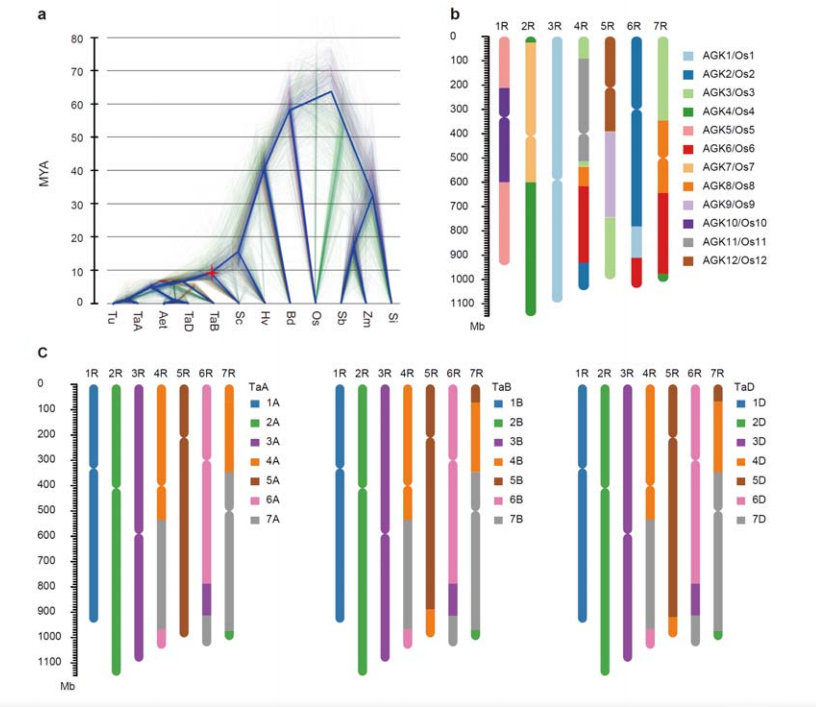
Evolutions- und Chromosomensyntenieanalysen des Roggengenoms
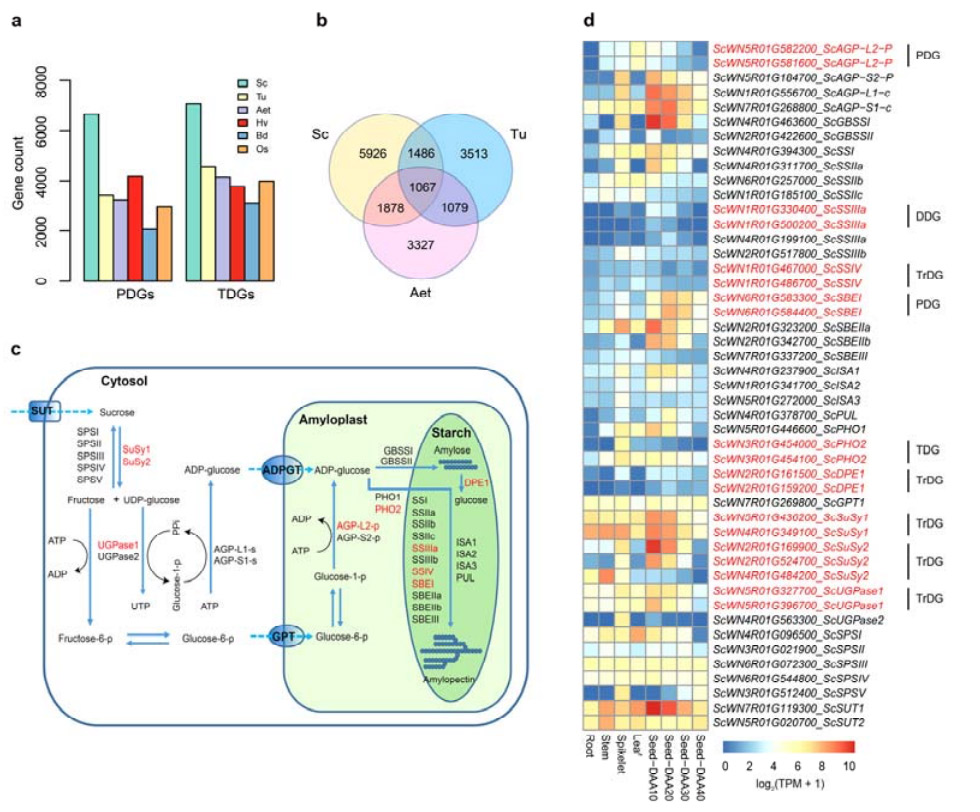
Analyse von Roggen-Genduplikationen und deren Einfluss auf die Diversität von Stärkebiosynthese-bezogenen Genen (SBRGs)
5. Präparation der Genorte des Roggensamenspeicherproteins (SSP).
Auf 1R oder 2R wurden vier chromosomale Loci (Sec-1 bis Sec-4) identifiziert, die Roggen-SSPs spezifizieren.α-Gliadin-Gene wurden erst kürzlich in Weizen und eng verwandten Arten nach der Divergenz von Weizen und Roggen entwickelt.
6. Untersuchung des Transkriptionsfaktors (TF) und der Krankheitsresistenzgene
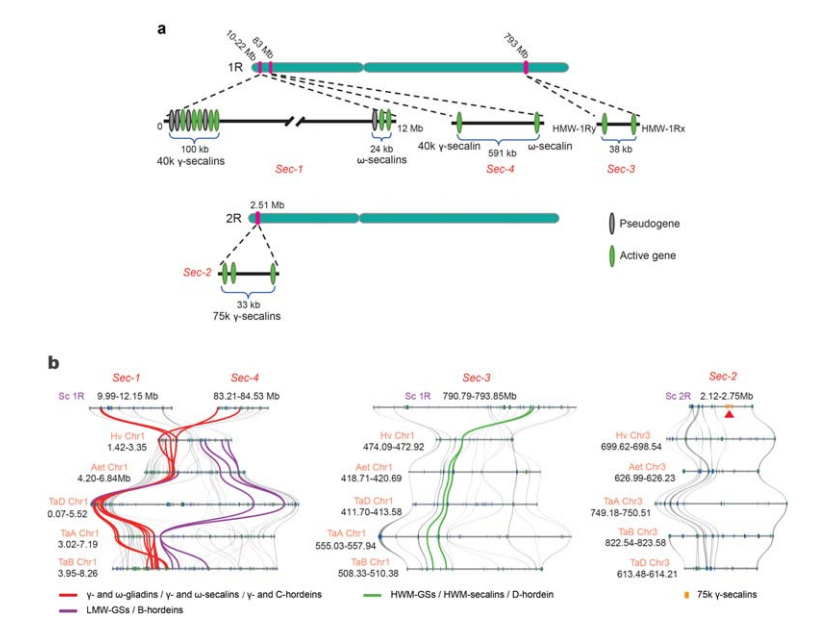
Analyse von Roggen-Secalin-Loci
Weining-Roggen hatte zahlreichere Krankheitsresistenz-assoziierte (DRA) Gene (1.989, Ergänzende Daten 3) als Tu (1.621), Aet (1.758), Hv (1.508), Bd (1.178), Os (1.575) und A (1.836). ), B (1.728) und D (1.888) Subgenome von Weichweizen.
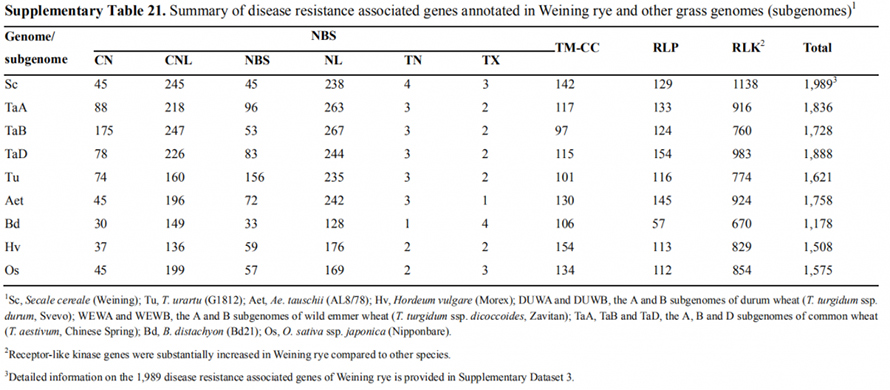
7. Untersuchung von Genexpressionsmerkmalen, die mit dem frühen Kopfmerkmal verbunden sind
Zwei FT-Gene mit relativ hoher Expression unter Langtagbedingungen, ScFT1 und ScFT2, wurden bei der Weining-Genomassemblierung annotiert.Bei zwei Aminosäureresten der ScFT2-Phosphorylierung (S76 und T132) wurde ein Zusammenhang mit der Verringerung der Zeitkontrolle gefunden
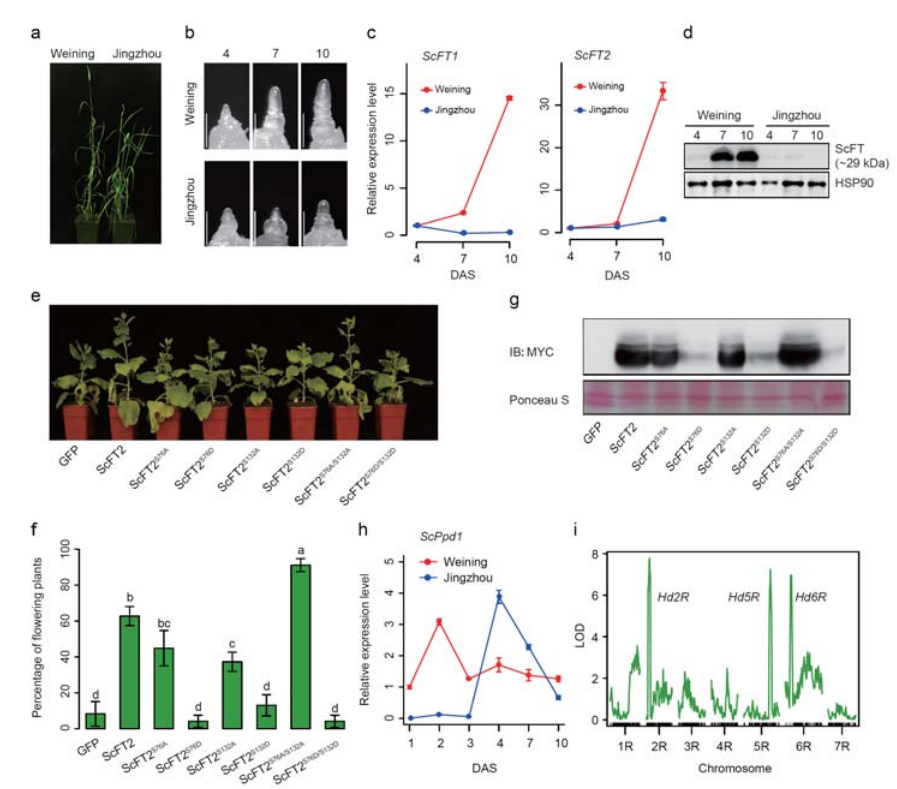
Entwicklungs- und Genexpressionsmerkmale, die mit dem frühen Triebmerkmal von Weining-Roggen verbunden sind
8. Abbau chromosomaler Regionen und Loci, die möglicherweise an der Domestizierung von Roggen beteiligt sind
Insgesamt 123.647 SNPs wurden verwendet, um eine selektive Sweep-Analyse zwischen Kulturroggen und S. vavilovii durchzuführen.11 selektive Sweep-Signale, identifiziert durch Reduktionsindex (DRI), Fixierungsindex (FST) und XP-CLR-Methode.Es wurde festgestellt, dass ScID1 möglicherweise an der Regulierung des Überschriftendatums beteiligt ist.
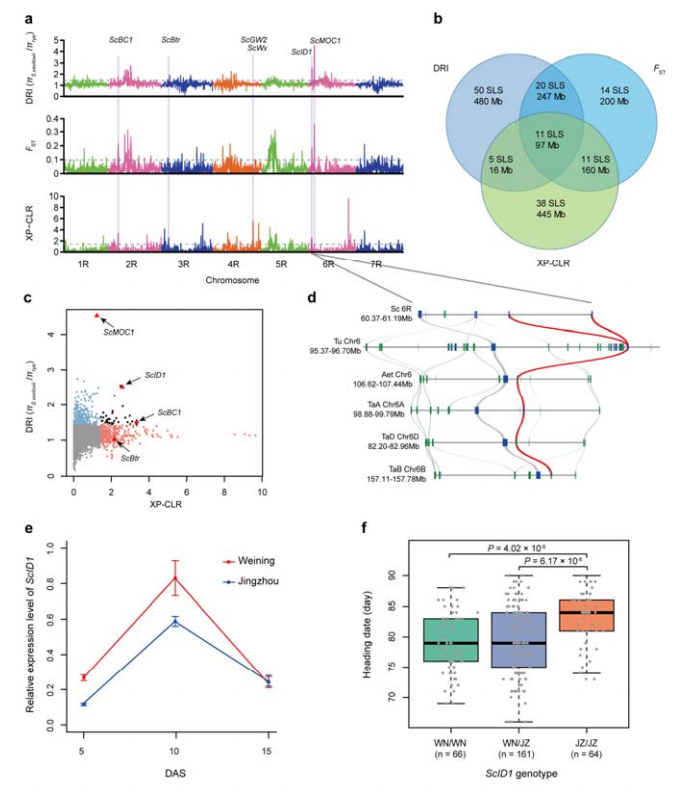
Identifizierung und Analyse chromosomaler Regionen und Loci, die möglicherweise mit der Domestizierung von Roggen in Zusammenhang stehen
Referenz
Li GW et al.Eine qualitativ hochwertige Genomassemblierung hebt die genomischen Merkmale und agronomisch wichtigen Gene des Roggens hervor.Naturgenetik (2021)
Neuigkeiten und Highlights Ziel ist es, die neuesten erfolgreichen Fälle mit Biomarker-Technologien zu teilen und neue wissenschaftliche Errungenschaften sowie herausragende Techniken, die während der Studie angewendet wurden, zu erfassen.
Zeitpunkt der Veröffentlichung: 05.01.2022