

Hi-C-basierte Genomassemblierung







Servicevorteile
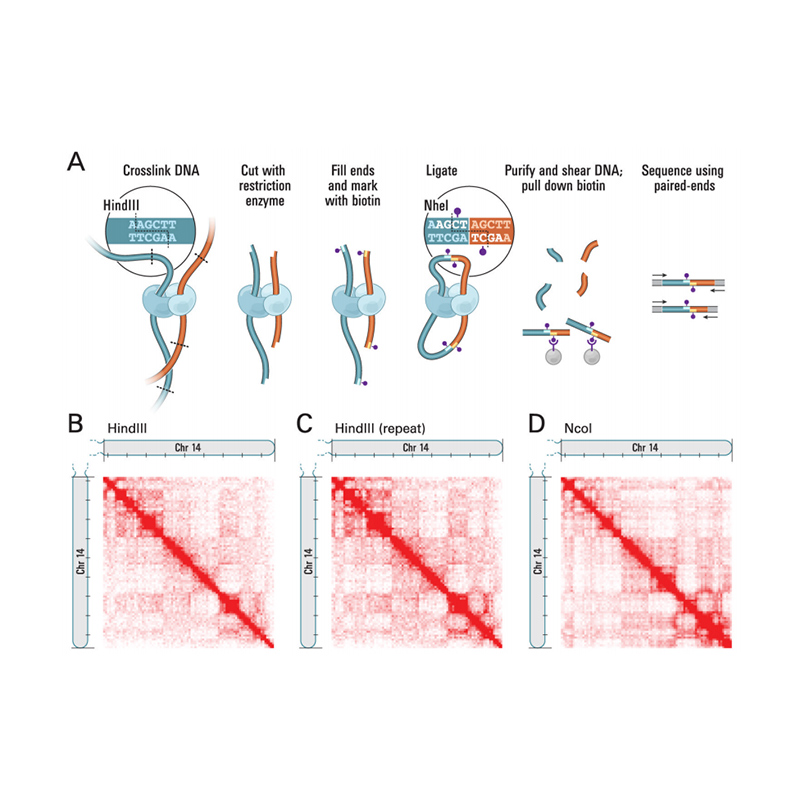
Übersicht über Hi-C
(Lieberman-Aiden E et al.,Wissenschaft, 2009)
● Es ist nicht erforderlich, eine genetische Population für die Contig-Verankerung zu konstruieren.
● Höhere Markierungsdichte führt zu einer höheren Verankerungsquote der Contigs von über 90 %;
● Ermöglicht die Bewertung und Korrektur vorhandener Genomassemblierungen;
● Kürzere Durchlaufzeiten mit höherer Genauigkeit bei der Genomassemblierung;
● Umfangreiche Erfahrung mit über 1000 Hi-C-Bibliotheken, die für über 500 Arten erstellt wurden;
● Über 100 erfolgreiche Fälle mit einem kumulativen veröffentlichten Impact Factor von über 760;
● Hi-C-basierte Genomassemblierung für polyploides Genom, 100 % Verankerungsrate wurde im vorherigen Projekt erreicht;
● Interne Patente und Software-Urheberrechte für Hi-C-Experimente und Datenanalysen;
● Selbstentwickelte Software zur Optimierung visualisierter Daten, die das manuelle Verschieben, Umkehren, Widerrufen und Wiederherstellen von Blöcken ermöglicht.
Leistungsbeschreibung
|
Bibliothekstyp
|
Plattform | Leselänge | Strategie empfehlen |
| Hi-C | Illumina NovaSeq | PE150 | ≥ 100X |
Bioinformatische Analysen
● Qualitätskontrolle der Rohdaten
● Qualitätskontrolle der Hi-C-Bibliothek
● Hi-C-basierte Genomassemblierung
● Bewertung nach der Montage
Musteranforderungen und Lieferung
Probenanforderungen:
| Tier | Pilz | Pflanzen
|
| Gefrorenes Gewebe: 1–2 g pro Bibliothek Zellen: 1x 10^7 Zellen pro Bibliothek | Gefrorenes Gewebe: 1 g pro Bibliothek | Gefrorenes Gewebe: 1–2 g pro Bibliothek
|
| *Wir empfehlen dringend, mindestens 2 Aliquots (jeweils 1 g) für das Hi-C-Experiment einzusenden. | ||
Empfohlene Musterlieferung
Behälter: 2 ml Zentrifugenröhrchen (Alufolie wird nicht empfohlen)
Für die meisten Proben empfehlen wir, sie nicht in Ethanol aufzubewahren.
Probenkennzeichnung: Proben müssen deutlich gekennzeichnet sein und mit dem eingereichten Probeninformationsformular identisch sein.
Versand: Trockeneis: Die Proben müssen zunächst in Beutel verpackt und in Trockeneis vergraben werden.
Service-Workflow

Experimentdesign

Musterlieferung

DNA-Extraktion

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
*Die hier gezeigten Demo-Ergebnisse stammen alle von Genomen, die mit Biomarker Technologies veröffentlicht wurden
1.Hi-C-Interaktionswärmekarte vonCamptotheca acuminataGenom.Wie auf der Karte dargestellt, korreliert die Intensität der Interaktionen negativ mit der linearen Entfernung, was auf eine hochpräzise Anordnung auf Chromosomenebene hinweist.(Verankerungsverhältnis: 96,03 %)
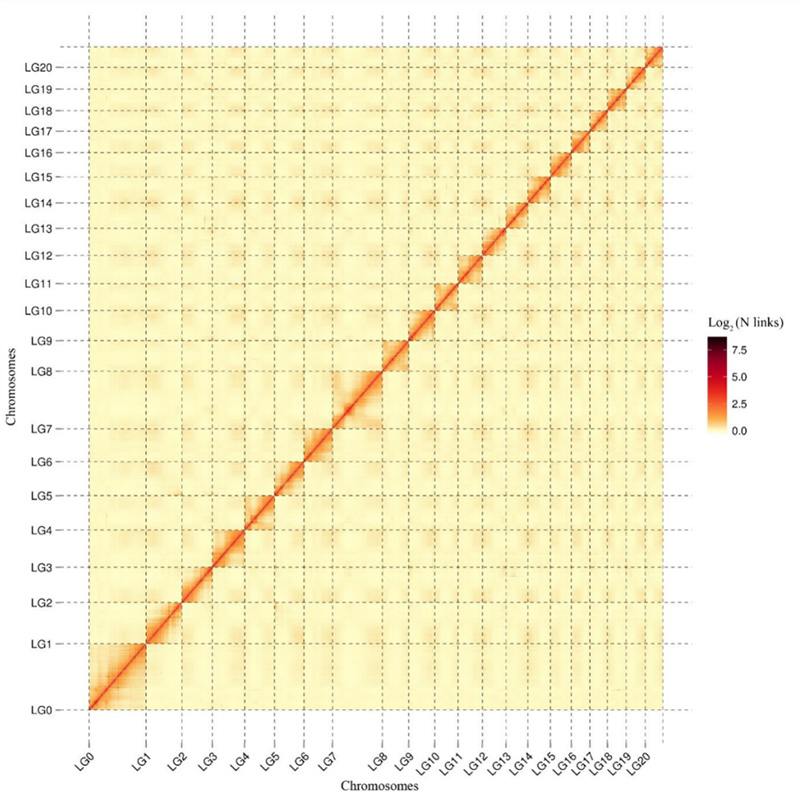
Kang M et al.,Naturkommunikation, 2021
2.Hi-C erleichterte die Validierung von Inversionen zwischenGossypium hirsutumL. TM-1 A06 undG. ArboreumChr06
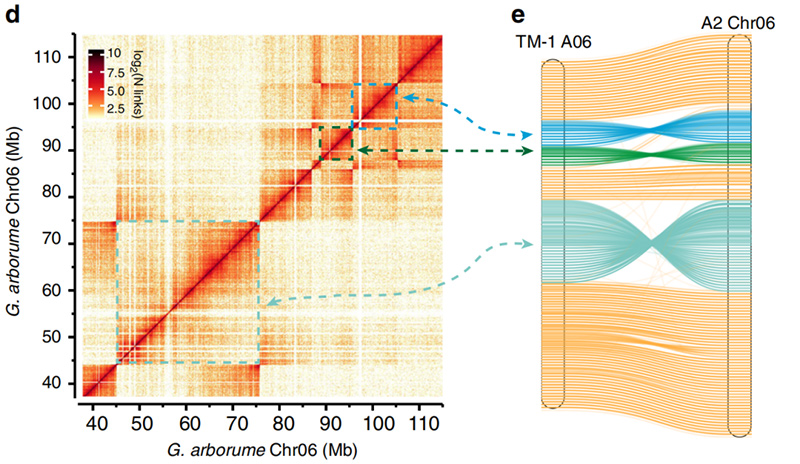
Yang Z et al.,Naturkommunikation, 2019
3.Assemblierung und biallelische Differenzierung des Cassava-Genoms SC205.Die Hi-C-Heatmap zeigt eine deutliche Aufteilung homologer Chromosomen.
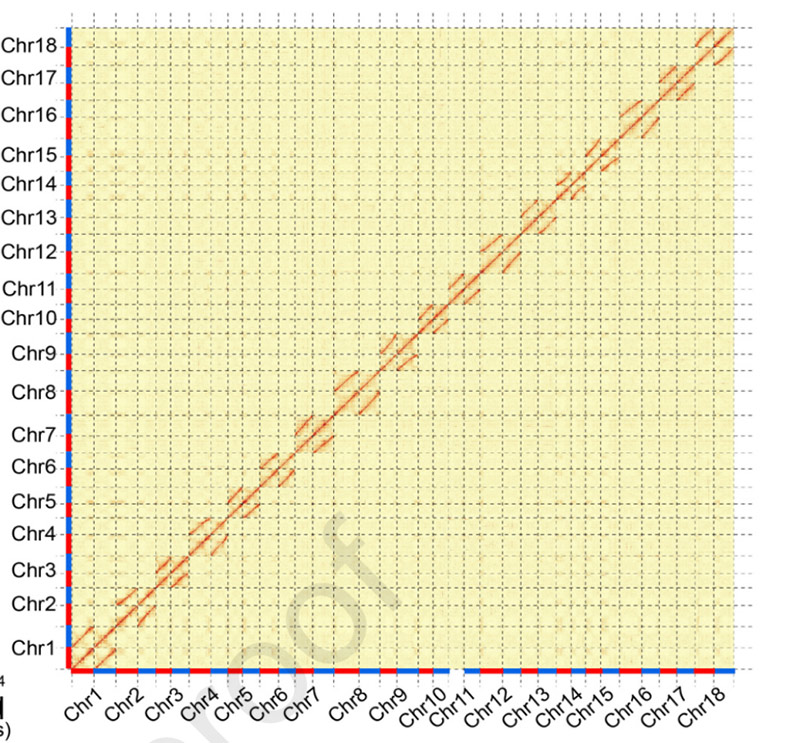
Hu W et al.,Molekulare Pflanze, 2021
4.Hi-C-Heatmap auf dem Genomaufbau zweier Ficus-Arten:F.microcarpa(Verankerungsverhältnis: 99,3 %) undF.hispida (Verankerungsverhältnis: 99,7 %)
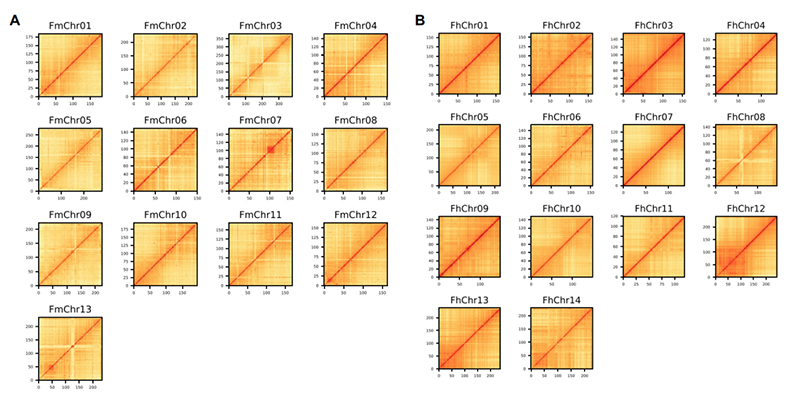
Zhang X et al.,Zelle, 2020
BMK-Fall
Genome des Banyanbaums und der Bestäuberwespe geben Einblicke in die Koevolution von Feigenwespe
Veröffentlicht: Zelle, 2020
Sequenzierungsstrategie:
F. microcarpa Genom: Ca.84 x PacBio RSII (36,87 GB) + Hi-C (44 GB)
F. hispidaGenom: Ca.97 x PacBio RSII (36,12 GB) + Hi-C (60 GB)
Eupristina verticillataGenom: Ca.170 x PacBio RSII (65 GB)
Wichtigste Ergebnisse
1.Zwei Banyan-Tree-Genome und ein Bestäuberwespengenom wurden mithilfe von PacBio-Sequenzierung, Hi-C und Verknüpfungskarte konstruiert.
(1)F. microcarpaGenom: Es wurde eine Anordnung von 426 MB (97,7 % der geschätzten Genomgröße) mit Contig N50 von 908 Kb und einem BUSCO-Score von 95,6 % erstellt.Insgesamt wurden 423 Mb-Sequenzen durch Hi-C auf 13 Chromosomen verankert.Die Annotation des Genoms ergab 29.416 proteinkodierende Gene.
(2)F. HispidaGenom: Eine Anordnung von 360 MB (97,3 % der geschätzten Genomgröße) ergab einen Ertrag mit einem Contig N50 von 492 Kb und einem BUSCO-Score von 97,4 %.Insgesamt 359 Mb-Sequenzen wurden durch Hi-C auf 14 Chromosomen verankert und waren mit der High-Density-Linkage-Map äußerst identisch.
(3)Eupristina verticillataGenom: Es wurde eine Anordnung von 387 MB (geschätzte Genomgröße: 382 MB) mit einem Contig-N50 von 3,1 MB und einem BUSCO-Score von 97,7 % erstellt.
2. Eine vergleichende Genomanalyse ergab eine große Anzahl von Strukturvariationen zwischen zweiFicusGenomen, die eine unschätzbare genetische Ressource für Studien zur adaptiven Evolution darstellten.Diese Studie lieferte zum ersten Mal Einblicke in die Koevolution von Feigenwespen auf genomischer Ebene.
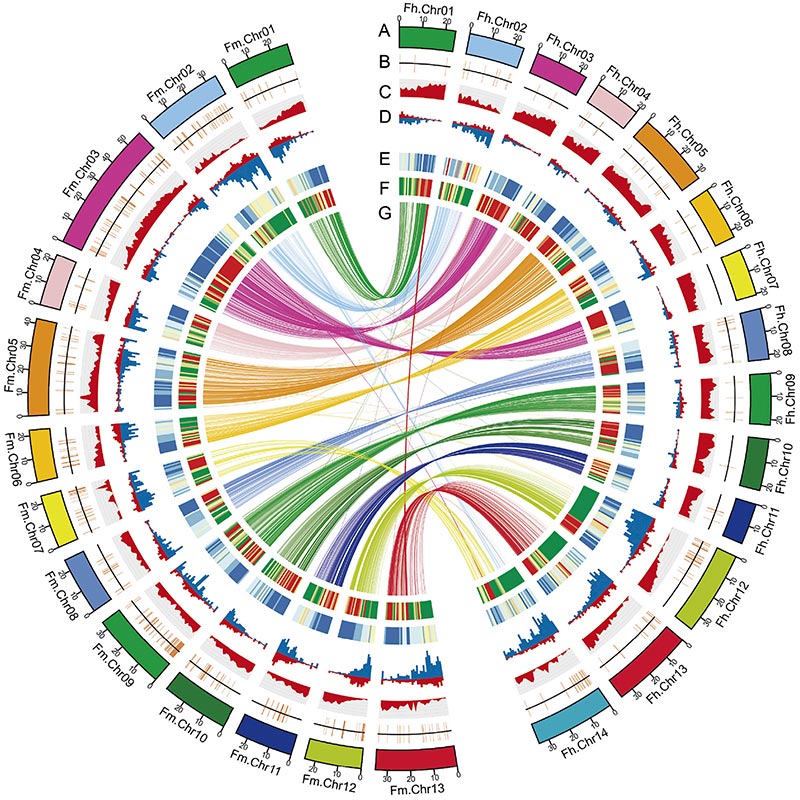 Circos-Diagramm zu genomischen Merkmalen von zweiFicusGenome, einschließlich Chromosomen, segmentale Duplikationen (SDs), Transposons (LTR, TEs, DNA TEs), Genexpression und Syntenie | 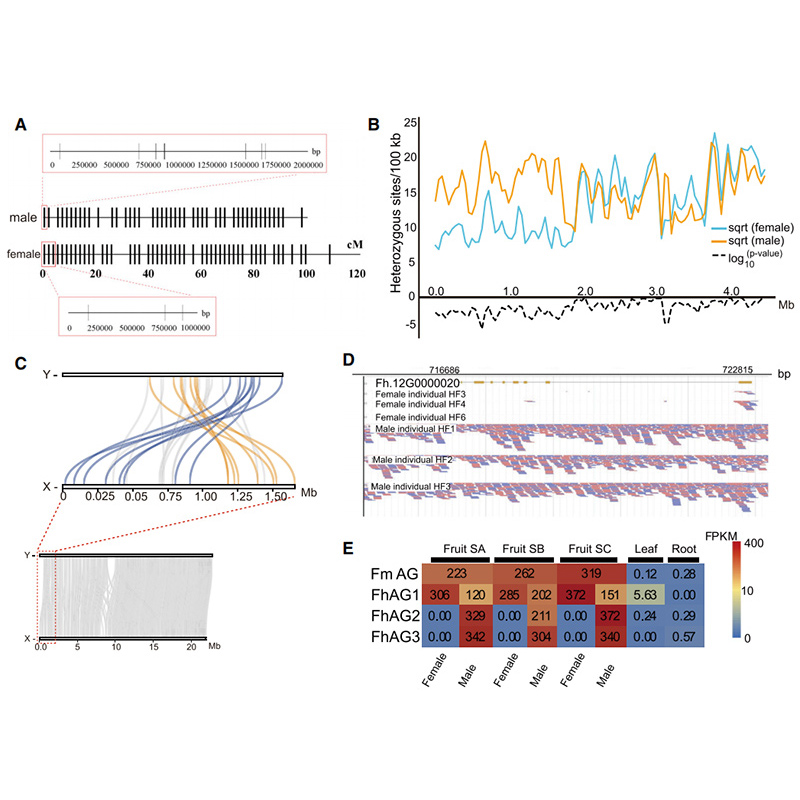 Identifizierung des Y-Chromosoms und Kandidatengen zur Geschlechtsbestimmung |
Zhang, X., et al.„Genome des Banyanbaums und der Bestäuberwespe bieten Einblicke in die Koevolution von Feigenwespe.“Zelle 183,4 (2020).





