

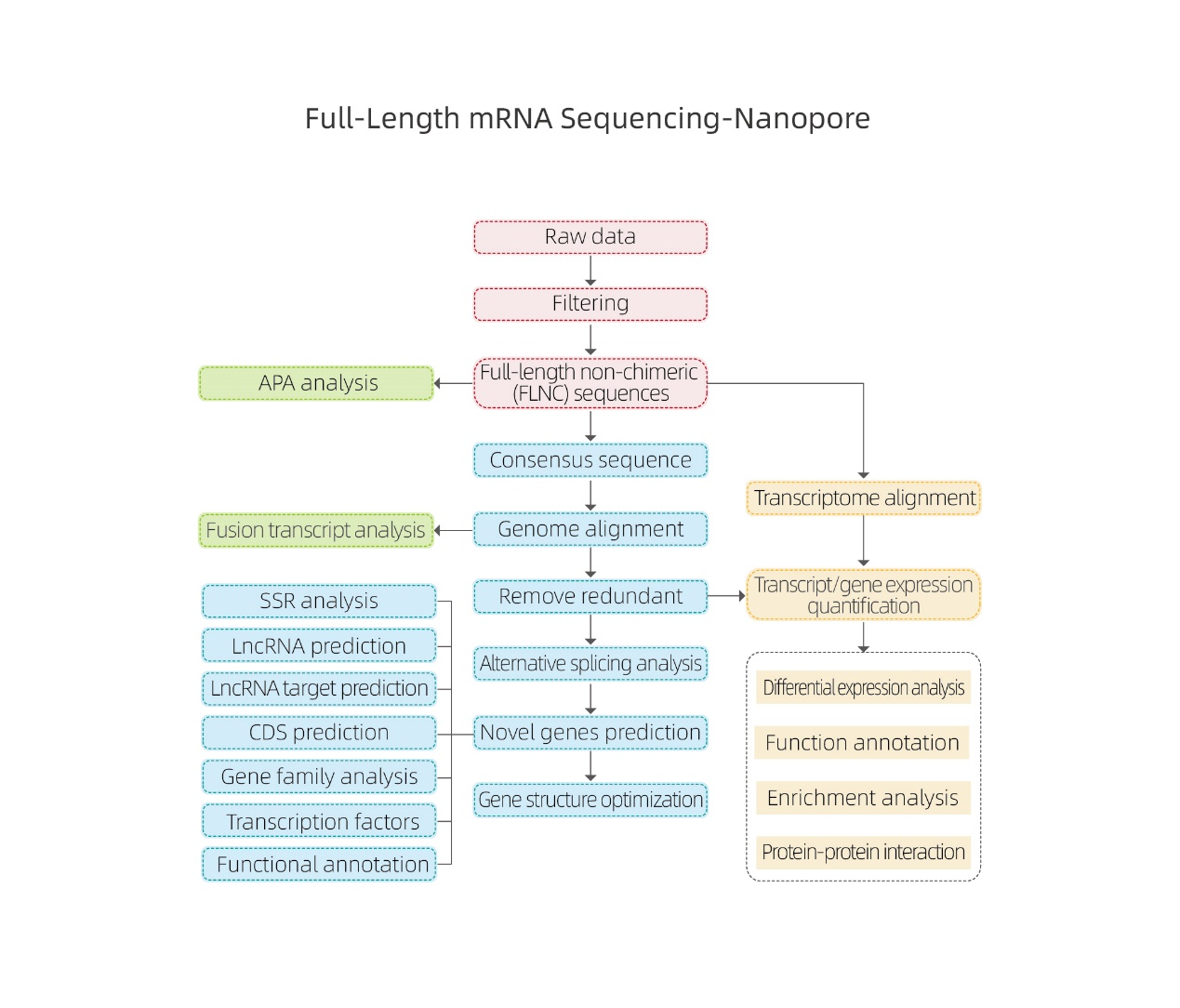
Vollständige mRNA-Sequenzierung – Nanopore





Servicevorteile
● Geringe Sequenzverzerrung
● Offenlegung von cDNA-Molekülen voller Länge
● Weniger Daten erforderlich, um die gleiche Anzahl von Transkripten abzudecken
● Identifizierung mehrerer Isoformen pro Gen
● Expressionsquantifizierung auf Isoformenebene
Leistungsbeschreibung
| Bibliothek | Plattform | Empfohlener Datenertrag (GB) | Qualitätskontrolle |
| cDNA-PCR (Poly-A angereichert) | Nanopore PromethION P48 | 6 GB/Probe (abhängig von der Art) | Verhältnis in voller Länge > 70 % Durchschnittlicher Qualitätsfaktor: Q10
|
Bioinformatische Analysen
●Rohdatenverarbeitung
● Transkriptidentifizierung
● Alternatives Spleißen
● Expressionsquantifizierung auf Genebene und Isoformenebene
● Differentialausdrucksanalyse
● Funktionsannotation und -anreicherung (DEGs und DETs)
Musteranforderungen und Lieferung
Probenanforderungen:
Nukleotide:
| Konz. (ng/μl) | Menge (μg) | Reinheit | Integrität |
| ≥ 100 | ≥ 0,6 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Begrenzte oder keine Protein- oder DNA-Kontamination auf dem Gel sichtbar. | Für Pflanzen: RIN≥7,0; Für Tiere: RIN≥7,5; 5,0≥28S/18S≥1,0; begrenzte oder keine Grundlinienhöhe |
Gewebe: Gewicht (trocken): ≥1 g
*Für Gewebe mit weniger als 5 mg empfehlen wir, schockgefrorene (in flüssigem Stickstoff) Gewebeproben einzusenden.
Zellsuspension: Zellzahl = 3×106- 1×107
*Wir empfehlen den Versand von gefrorenem Zelllysat.Für den Fall, dass die Zellenzahl kleiner als 5×10 ist5Es wird empfohlen, in flüssigem Stickstoff schockgefrostet zu werden, was für die Mikroextraktion vorzuziehen ist.
Blutproben: Volumen ≥ 1 ml
Empfohlene Musterlieferung
Behälter: 2 ml Zentrifugenröhrchen (Alufolie wird nicht empfohlen)
Probenbeschriftung: Gruppe+Replikation, z. B. A1, A2, A3;B1, B2, B3... ...
Versand: 2、Trockeneis: Die Proben müssen in Beutel verpackt und in Trockeneis vergraben werden.
- RNAstable-Röhrchen: RNA-Proben können in RNA-Stabilisierungsröhrchen (z. B. RNAstable®) getrocknet und bei Raumtemperatur versendet werden.
Service-Workflow
Nukleotide:

Musterlieferung

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
Service-Workflow
Gewebe:

Experimentdesign
Musterlieferung

RNA-Extraktion
Bibliotheksbau
Sequenzierung
Datenanalyse
Kundendienst
1.Differentialausdrucksanalyse – Vulkandiagramm
Die Analyse der differentiellen Expression kann sowohl auf Genebene zur Identifizierung differenziell exprimierter Gene (DEGs) als auch auf Isoformenebene zur differenziellen Identifizierung durchgeführt werden
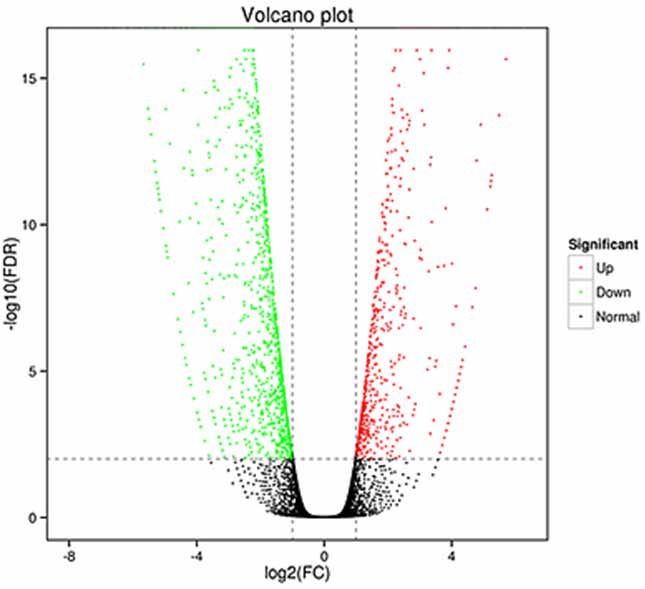
ausgedrückte Transkripte (DETs)
2.Hierarchische Clustering-Heatmap

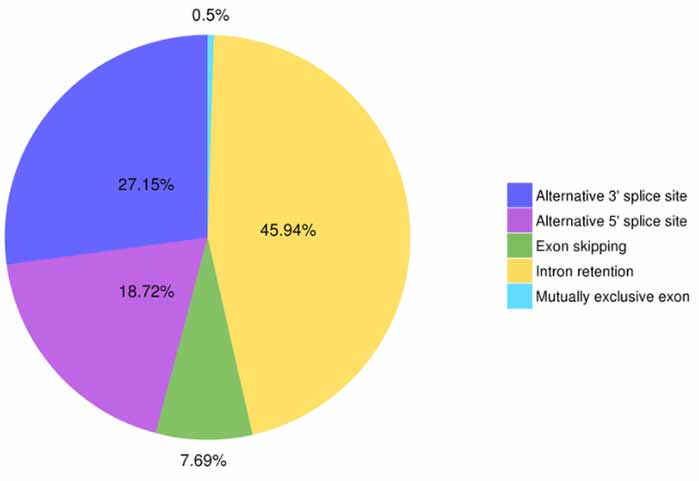
3. Identifizierung und Klassifizierung alternativer Spleißverbindungen
Fünf Arten alternativer Spleißereignisse können von Astalavista vorhergesagt werden.
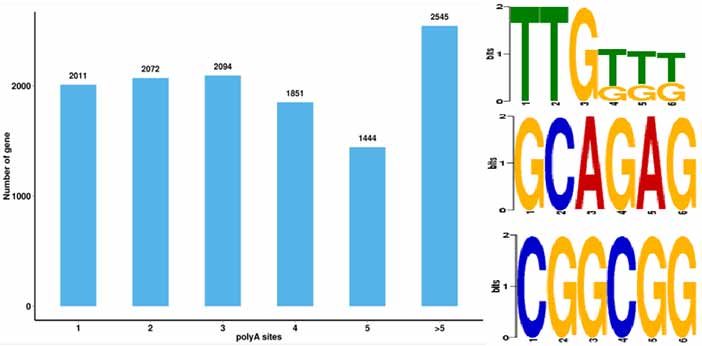
4.Identifizierung alternativer Polyadenylierungsereignisse (APA) und Motiv 50 bp stromaufwärts von Poly-A
BMK-Fall
Alternative Spleißidentifizierung und Quantifizierung auf Isoformebene durch Nanoporen-Transkriptomsequenzierung in voller Länge
Veröffentlicht:Naturkommunikation, 2020
Sequenzierungsstrategie:
Gruppierung: 1. CLL-SF3B1(WT);2. CLL-SF3B1 (K700E-Mutation);3. Normale B-Zellen
Sequenzierungsstrategie: MinION 2D-Bibliothekssequenzierung, PromethION 1D-Bibliothekssequenzierung;Kurzlesen von Daten aus denselben Proben
Sequenzierungsplattform: Nanopore MinION;Nanoporen-PromethION;
Wichtigste Ergebnisse
1. Alternative Spleißidentifizierung auf Isoform-Ebene
Lang gelesene Sequenzen ermöglichen die Identifizierung des mutierten SF3B1K700E-veränderte Spleißstellen auf Isoform-Ebene.Es wurde festgestellt, dass 35 alternative 3'SSs und 10 alternative 5'SSs zwischen SF3B1 signifikant unterschiedlich gespleißt warenK700Eund SF3B1WT.33 der 35 Veränderungen wurden durch lange Lesesequenzen neu entdeckt.
2. Alternative Splicing-Quantifizierung auf Isoform-Ebene
Expression von Intron-Retention(IR)-Isoformen in SF3B1K700Eund SF3B1WTwurden anhand von Nanoporensequenzen quantifiziert und zeigten eine globale Herunterregulierung der IR-Isoformen in SF3B1K700E.
Referenz
Tang AD, Soulette CM, Baren MJV, et al.Die vollständige Transkriptcharakterisierung der SF3B1-Mutation bei chronischer lymphatischer Leukämie zeigt eine Herunterregulierung zurückgehaltener Introns[J].Naturkommunikation.





