

Evolutionäre Genetik








Servicevorteile
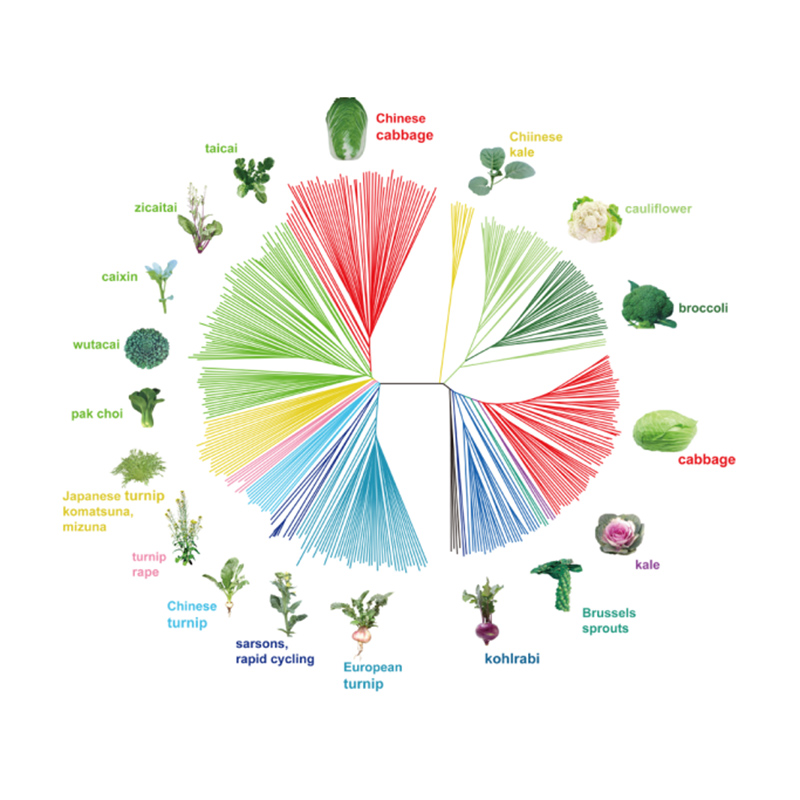
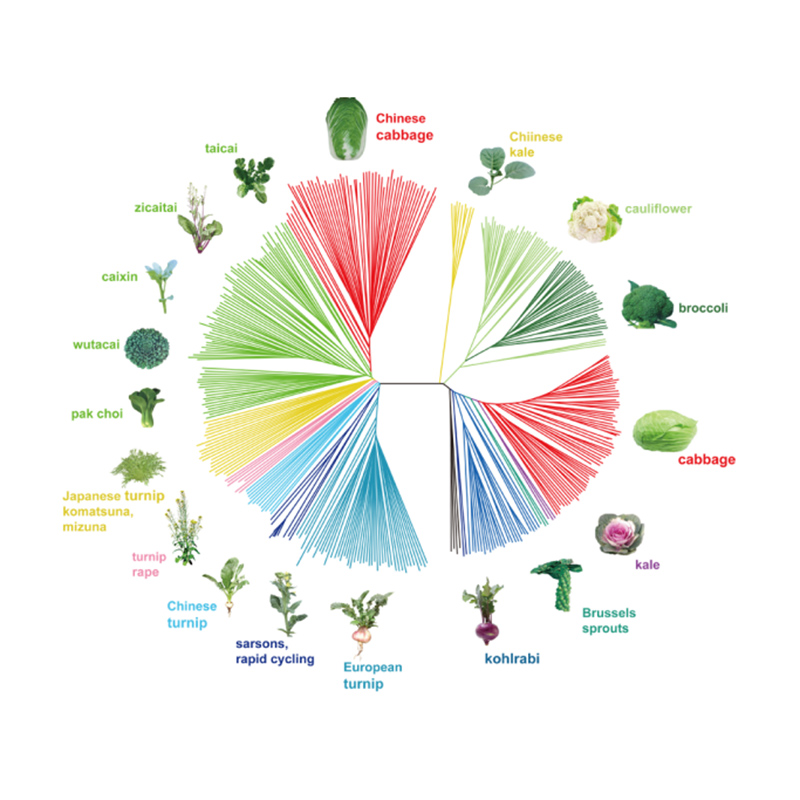
Takagi et al.,Das Pflanzenjournal, 2013
● Schätzung der Zeit und Geschwindigkeit der Artendivergenz basierend auf Variationen auf Nukleotid- und Aminosäureebene
● Aufdeckung zuverlässigerer phylogenetischer Beziehungen zwischen Arten mit minimiertem Einfluss konvergenter und paralleler Evolution
● Herstellung von Verbindungen zwischen genetischen Veränderungen und Phänotypen, um merkmalsbezogene Gene aufzudecken
● Abschätzung der genetischen Vielfalt, die das evolutionäre Potenzial von Arten widerspiegelt
● Schnellere Bearbeitungszeit
● Umfangreiche Erfahrung: BMK hat über 12 Jahre lang umfangreiche Erfahrung in bevölkerungs- und evolutionsbezogenen Projekten gesammelt, die Hunderte von Arten usw. abdecken, und an über 80 hochrangigen Projekten mitgewirkt, die in Nature Communications, Molecular Plants, Plant Biotechnology Journal usw. veröffentlicht wurden.
Leistungsbeschreibung
Materialien:
Normalerweise werden mindestens drei Subpopulationen (z. B. Unterarten oder Stämme) empfohlen.Jede Subpopulation sollte nicht weniger als 10 Individuen umfassen (Pflanzen > 15, kann bei seltenen Arten reduziert werden).
Sequenzierungsstrategie:
* WGS kann für Arten mit qualitativ hochwertigem Referenzgenom eingesetzt werden, während SLAF-Seq auf Arten mit oder ohne Referenzgenom oder auf Referenzgenome von schlechter Qualität anwendbar ist.
| Anwendbar auf die Genomgröße | WGS | SLAF-Tags (×10.000) |
| ≤ 500 MB | 10×/Einzelperson | WGS ist eher zu empfehlen |
| 500 MB – 1 GB | 10 | |
| 1 GB - 2 GB | 20 | |
| ≥2 GB | 30 |
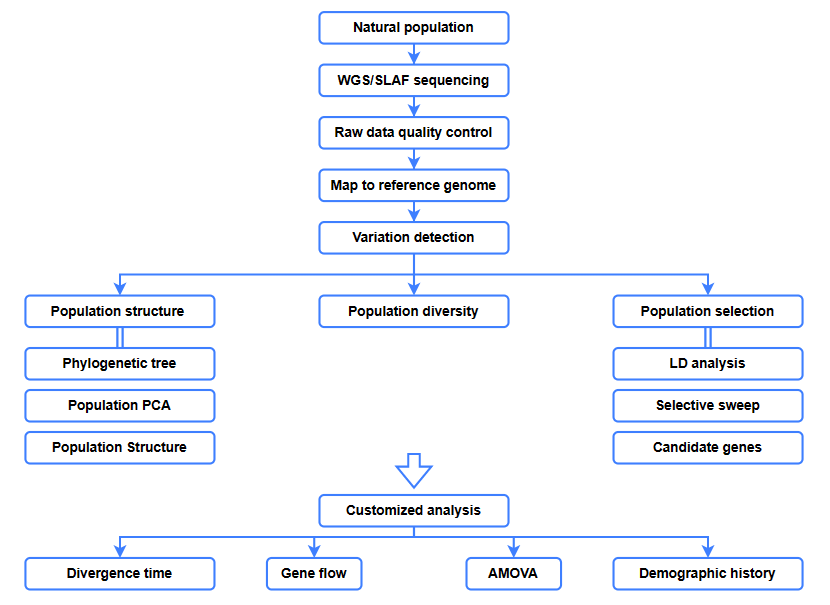
Bioinformatische Analysen
● Evolutionsanalyse
● Selektiver Sweep
● Genfluss
● Demografische Geschichte
● Divergenzzeit
Musteranforderungen und Lieferung
Probenanforderungen:
| Spezies | Gewebe | WGS-NGS | SLAF |
| Tier
| Viszerales Gewebe |
0,5~1g
|
0,5g
|
| Muskelgewebe | |||
| Säugetierblut | 1,5 ml
| 1,5 ml
| |
| Geflügel-/Fischblut | |||
| Anlage
| Frisches Blatt | 1~2g | 0,5~1g |
| Blütenblatt/Stiel | |||
| Wurzel/Samen | |||
| Zellen | Kultivierte Zelle |
| gDNA | Konzentration | Menge (ug) | OD260/OD280 |
| SLAF | ≥35 | ≥1,6 | 1,6-2,5 |
| WGS-NGS | ≥1 | ≥0,1 | - |
Service-Workflow

Experimentdesign

Musterlieferung

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
*Die hier gezeigten Demo-Ergebnisse stammen alle von mit BMKGENE veröffentlichten Genomen
1. Die Evolutionsanalyse umfasst die Konstruktion eines Stammbaums, einer Populationsstruktur und einer PCA auf der Grundlage genetischer Variationen.
Der phylogenetische Baum stellt taxonomische und evolutionäre Beziehungen zwischen Arten mit gemeinsamen Vorfahren dar.
PCA zielt darauf ab, die Nähe zwischen Teilpopulationen zu visualisieren.
Die Populationsstruktur zeigt das Vorhandensein einer genetisch unterschiedlichen Subpopulation hinsichtlich der Allelhäufigkeiten.
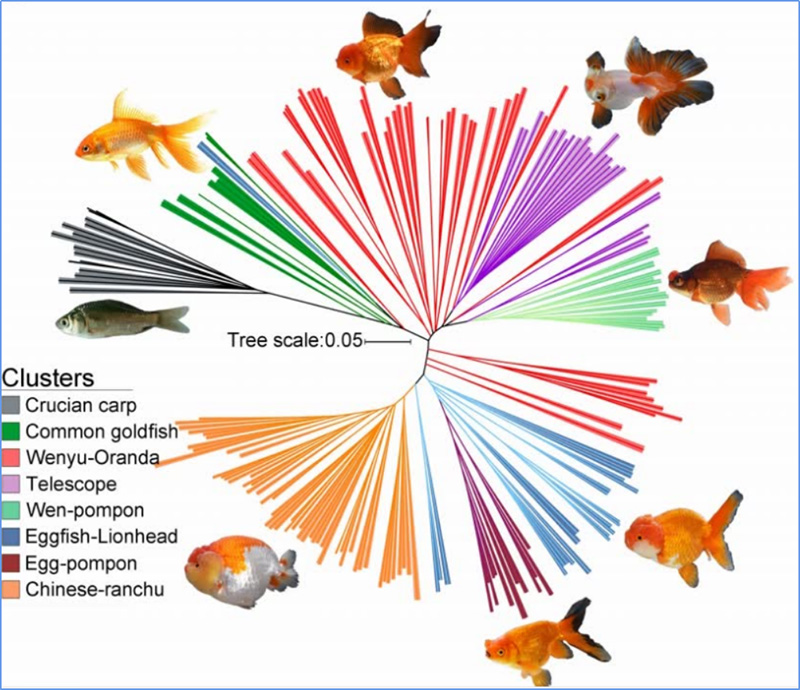
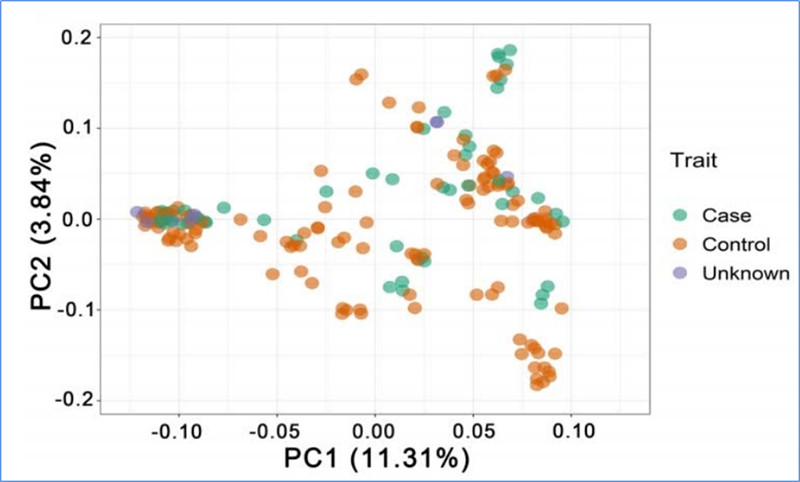
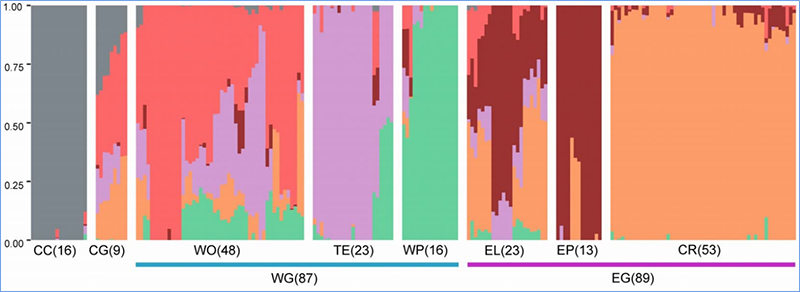
Chen et al.al.,PNAS, 2020
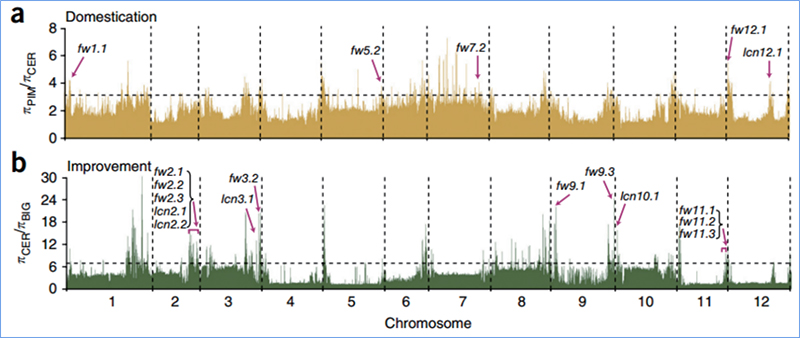
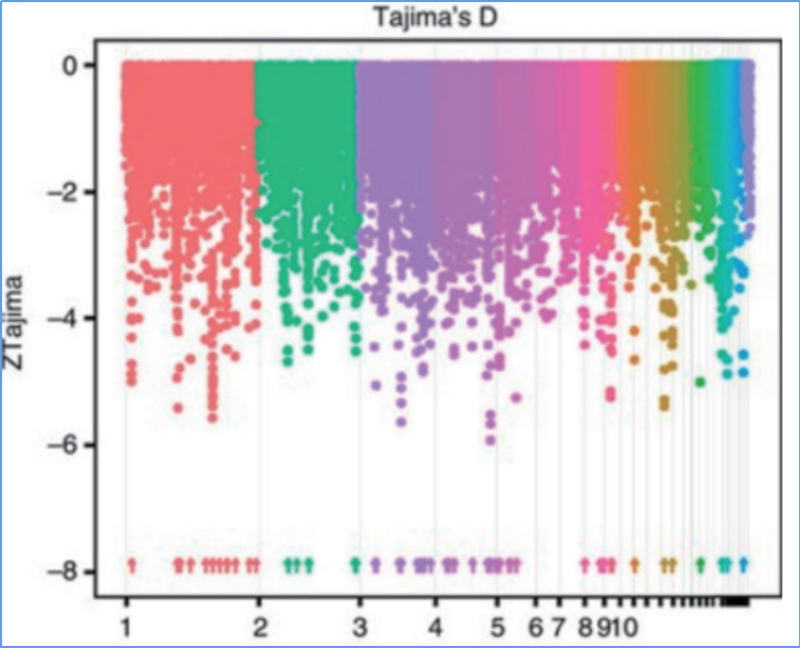
2. Selektiver Sweep
Unter selektivem Sweep versteht man einen Prozess, bei dem ein vorteilhafter Standort ausgewählt wird und die Häufigkeit verknüpfter neutraler Standorte erhöht und die Häufigkeit nicht verknüpfter Standorte verringert wird, was zu einer Verringerung regionaler Standorte führt.
Der genomweite Nachweis in selektiven Sweep-Regionen erfolgt durch Berechnung des populationsgenetischen Index (π,Fst, Tajima's D) aller SNPs innerhalb eines Schiebefensters (100 Kb) in einem bestimmten Schritt (10 Kb).
Nukleotidvielfalt(π)
Tajimas D
Fixationsindex (Fst)
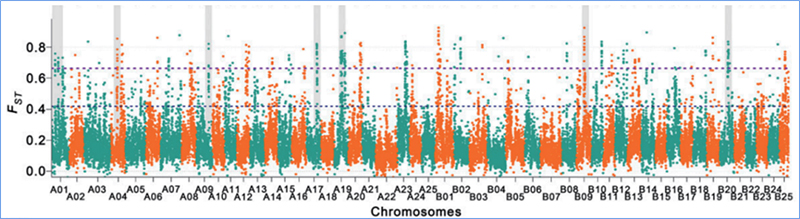
Wu, et.al.,Molekulare Pflanze, 2018
3.Genfluss
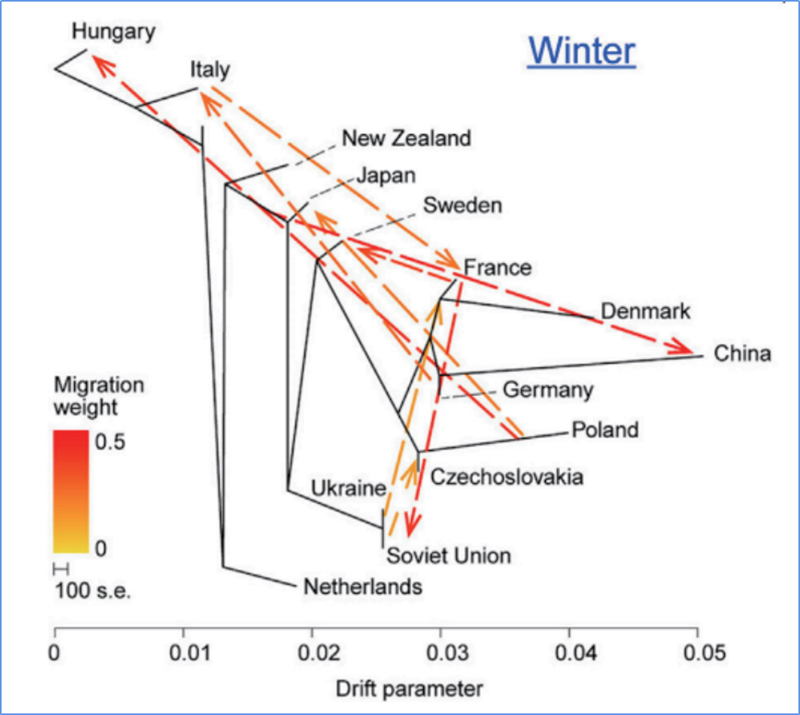
Wu, et.al.,Molekulare Pflanze, 2018
4.Demografische Geschichte
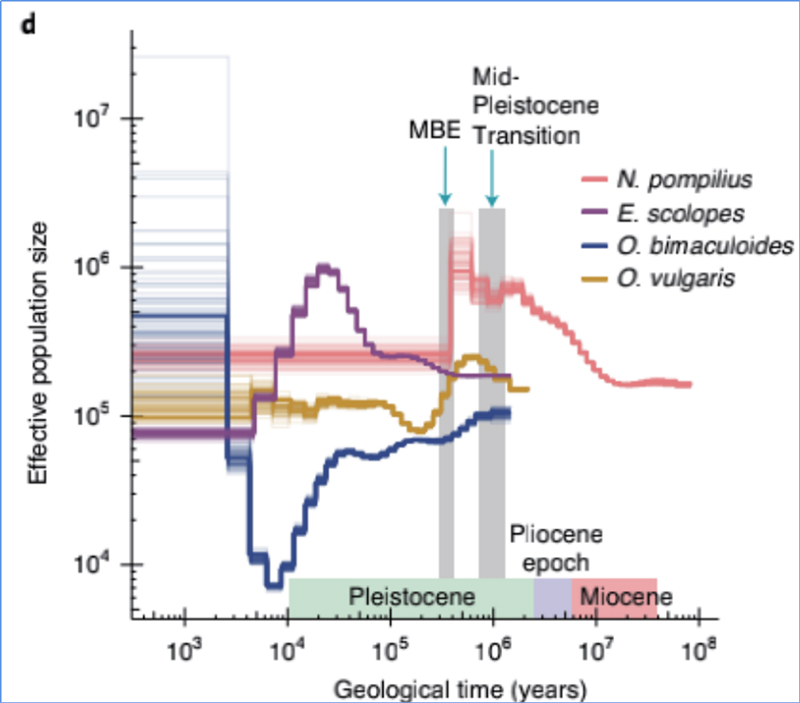
Zhang et al.al.,Naturökologie und Evolution, 2021
5.Divergenzzeit
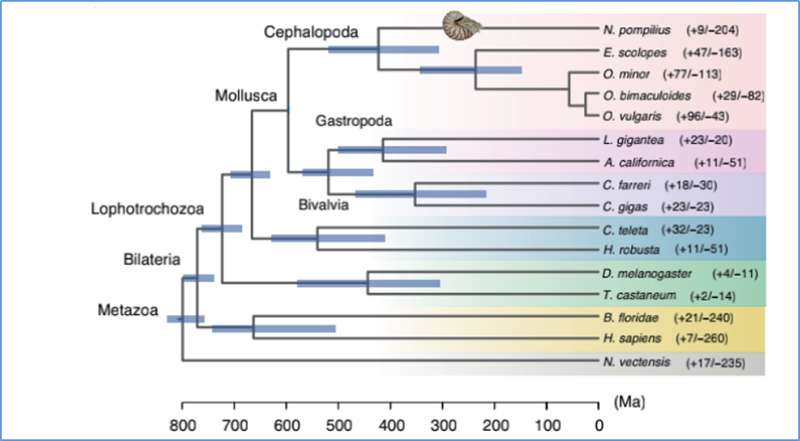
Zhang et al.al.,Naturökologie und Evolution, 2021
BMK-Fall
Eine genomische Variationskarte bietet Einblicke in die genetischen Grundlagen der Selektion von Frühlings-Chinakohl (Brassica rapa ssp. Pekinensis).
Veröffentlicht: Molekulare Pflanze, 2018
Sequenzierungsstrategie:
Neusequenzierung: Sequenzierungstiefe: 10×
Wichtigste Ergebnisse
In dieser Studie wurden 194 Chinakohlsorten für die Neusequenzierung mit einer durchschnittlichen Tiefe von 10x verarbeitet, was 1.208.499 SNPs und 416.070 InDels ergab.Die phylogenetische Analyse dieser 194 Linien ergab, dass diese Linien in drei Ökotypen unterteilt werden können: Frühling, Sommer und Herbst.Darüber hinaus deuteten die Populationsstruktur und die PCA-Analyse darauf hin, dass Frühlings-Chinakohl aus einem Herbstkohl in Shandong, China, stammte.Diese wurden anschließend in Korea und Japan eingeführt, mit lokalen Linien gekreuzt und einige spätblühende Sorten davon wurden wieder in China eingeführt und bildeten schließlich Frühlings-Chinakohl.
Das genomweite Scannen von Frühlings-Chinakohl und Herbstkohl bei der Selektion ergab 23 genomische Loci, die einer starken Selektion unterzogen wurden, von denen sich zwei mit der Region zur Steuerung der Schoßzeit basierend auf der QTL-Kartierung überlappten.Es wurde festgestellt, dass diese beiden Regionen Schlüsselgene enthalten, die die Blüte regulieren: BrVIN3.1 und BrFLC1.Durch Transkriptomstudien und transgene Experimente wurde außerdem bestätigt, dass diese beiden Gene an der Schoßzeit beteiligt sind.
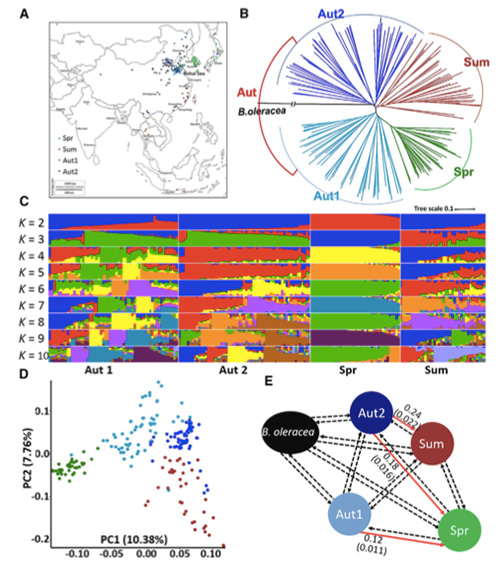 Analyse der Populationsstruktur von Chinakohl | 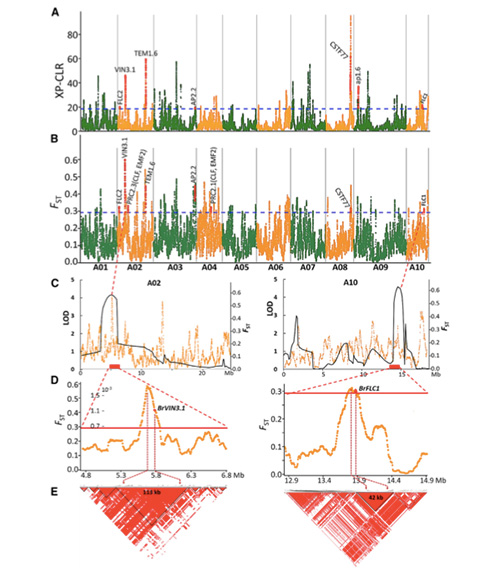 Genetische Informationen zur Auswahl von Chinakohl |
Tongbing et al.„Eine genomische Variationskarte bietet Einblicke in die genetischen Grundlagen der Selektion von Frühlings-Chinakohl (Brassica rapa ssp.pekinensis).“Molekulare Pflanzen,11(2018):1360-1376.





